As a medicine, valproic acid (VPA) has a variety of uses. Not only does it serve as an antiepileptic drug, it can also be used for mood stabilization in Bipolar, and has even found more recent use as an anti-cancer agent for certain types of tumors [1]. While it has already been well-established as a teratogen (a promoter of certain birth defects) there is a small body of literature that suggests VPA may also promote DNA mutations in those people taking the drug and in the offspring of mothers taking it during pregnancy [2 & 3, for examples].
The primary finding in these studies has been one of increased sister chromatid exchange (SCE). As is probably implied by the name, SCE is the exchange of genetic material between sister chromatids. (Wow, did that sound as redundant as I think it did? Anyways, see the image below.) This often occurs when a single chromatid is damaged and the missing material is then duplicated and supplied by the other chromatid. This can lead to mutation through repair because the homologous allele frequently doesn’t contain the same sequence as in that of the damaged segment. Just picture your cells having to use the genetic material your mother gave you to repair damage to your father’s genes.
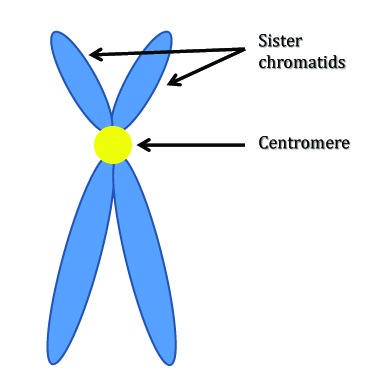
Double-stranded breaks are often a cause of SCE:

Valproic acid has a variety of downstream effects in tissues, but two of its direct effects are 1) blocking the synthesis of the carbohydrate, myo-inositol, a molecule integral to a number of second messenger systems within the cell, and 2) as a histone deacetylase (HDAC) inhibitor. For the purposes of mutegenicity this latter effect may be most important. HDACs function, as their name might imply, by deacetylating histones. Histones are proteins that aid in packing DNA into nucleosomes, a regulatory mechanism of gene transcription. As you can see from the image below a number of different histone proteins make up the larger octamer unit around which the DNA is wound.

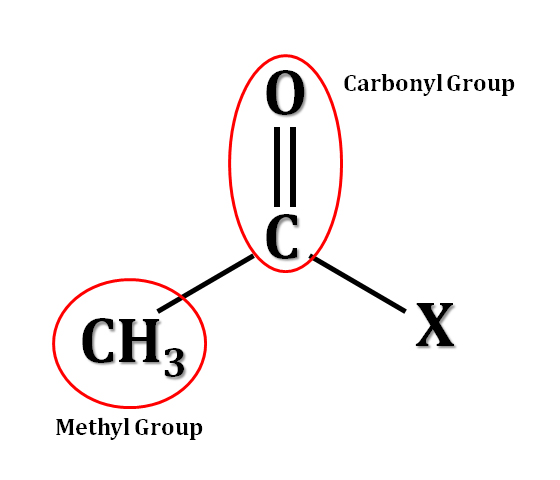
When a histone complex is acetylated, meaning that negatively-charged acetyl groups have been added to it, it loses some of its overall positive charge which decreases its capacity to interact with and bind tightly to DNA. This leaves the local DNA available for transcription (gene expression). Therefore, HDACs actively remove acetyl groups, allowing histones to better bind to DNA and maintain it in a closed form. Below you can see the chemical formula for an acetyl group which is comprised of a carbonyl group (a carbon double-bonded to an oxygen) and a methyl group; “X” is whatever the acetyl group happens to be bound to, in this case an amino acid, lysine, within the histone protein.
Why might VPA’s capacity to inhibit HDACs be so important when it comes to genome stability? I mean, hell, so the DNA’s a little unwound, maybe it’s a little more available for transcription. How could that make it less stable?
Well, there’s growing evidence to suggest that histone deacetylation is vital for not only maintaining genomic stability but also in repair processes [4, 5]. On average, the less “packaged” the DNA is, the more vulnerable it is to instability for a variety of reasons.
And so if we take this information and apply it to what we know about increased occurrences of sister chromatid exchange in cases of VPA exposure, it begins to make sense how a drug that inhibits HDAC activity could promote increased rates of certain types of mutations. We can expect those cells exposed to an HDAC inhibitor like valproic acid to be hyper-acetylated, in which the DNA is not tightly wound around the histone octamers and therefore genome stability is at higher risk.
Valproic acid has been an incredibly useful drug. For some epileptics and people with Bipolar it improves quality of life and makes their conditions more manageable; and for some people with cancer it’s no doubt even been a life saver. But I guess the takehome message of this story, aside from reminding the reader how truly delicate and mutable the genome is, is how delicate life itself can be. So be careful what you put in your body, because it’s the only one you’ve got.

I recently came across your blog via Dr. Casanova’s blog and will follow both of these interesting and informative blogs. The viability of the VPA autism mouse model has been confirmed by multiple independent research groups. I wasn’t aware, as you have pointed out, that exposure to VPA in pregnancy may have epigenetic effects in the womb and may disrupt gene expression. One of the most important relatively new findings in autism research has been the discovery of the significance of de novo gene mutations in autism. An increased rate of autism risk has been associated with de novo gene mutations both in the genetic syndromes and in simplex autism. The reason I ask this question is that all the VPA research has studied the effects of VPA on the offspring associated with exposure during pregnancy both in mice and in humans. I haven’t been able to find a study that looked at autism risk in women or mice that has studied the potential mutagenic effect of VPA exposure before conception.
Klinefelter Syndrome is the most common genetic syndrome that affects male newborns and occurs in 1 in 500 to 1,000 newborn males. Klinefelter Syndrome is not inherited and is always caused by an extra X chromosome. About half the cases are caused by XY sperm mutations and half the cases are caused by XX egg mutations.
http://ghr.nlm.nih.gov/condition/klinefelter-syndrome
Aside from Fragile X and a few very rare genetic syndromes almost all the children diagnosed with an unambiguous genetic syndrome are caused by reproductive cell errors (egg or sperm) and include Down Syndrome, Rett Syndrome, Prader-Willi Syndrome, Angelman Syndrome, Dup 12 duplication Syndrome, Phelan McDermid Syndrome, Timothy Syndrome and 22q11 deletion Syndrome to name a few associated with high autism risk.
Where do these reproductive errors come from? McCauliffe and colleagues examined the role of PCB and DDT congeners which still exist in the new classes of pesticides and the effect on sperm production in male volunteers recruited from a fertility clinic. What the study showed was that increasing levels of PCB and DDT congeners as measured in blood was associated with increased production of XY sperm mutations (Klinefelter Syndrome).
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3339457/
Marchetti’s group also discovered that workplace exposure to Benzene was associated with increased production of 1p36 sperm deletion mutations and is a risk factor for de novo 1p36 deletion syndrome one of the most devastating of the genetic syndromes:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3279447/
http://ghr.nlm.nih.gov/condition/1p36-deletion-syndrome
As long as the behavioral geneticists consider autism to be the most ‘heritable’ of all the developmental disorders research into this area will be left to evolutionary biologists and environmental health researchers.
Hi, Robert. You make a good point that there are no studies (of which I’m aware) that look at effects of VPA on mutation in germ cells, although I’m aware of studies that have looked at, for instance, sperm viability following large-dose VPA exposure: http://europepmc.org/abstract/MED/10845186
I should mention though that in order for a mutation to be seen throughout all organ systems, it of course needed to occur prior to or during gastrulation. If we’re talking epigenetic effects, then it either occurred in the germ cells prior to conception or occurred very early post-conception. BUT one could also see mutations in many but not all organ systems by targeting slightly later dates. Remember that the majority of autism mutations have been gauged, not by thorough investigation of all organ systems, but mainly by serum samples. I.e., we know these mutations occur in the blood cell progenitors (hematopoietic cells), so we could expect to see the same mutation in T-cells, B-cells, monocytes, macrophages, etc. But without being certain and biopsying the remainder of the organ systems, we really can’t say for certain that said mutation occurs throughout. If we did so, at the very least we could pinpoint different time periods if we’re talking exposure.
As you say, you haven’t been able to find a study that looks at pre-conception exposure to VPA and its effects on germ cells. I’m not myself aware of one. And it’s simply something that’s missing in the current literature and requires further investigation. At present, the trend I’ve noticed is that interest in potential mutagenic capacity of VPA has waned and probably isn’t being funded much. In the next couple of years, I have hopes of performing some in vitro experiments in these regards, in particular looking at more than simply double-stranded breaks.
Thanks for reading the blog, btw. I’m glad you found me through Manny’s blog. 🙂
Just a final thought, there is an assumption, certainly in the general population, that the genetic syndromes caused by reproductive errors (sperm or egg) are rare and random events. Not at all. Molina’s laboratory studied sperm mutations in 10 healthy male volunteer donors focusing on three mutations identified in individuals diagnosed with a genetic syndrome associated with high autism risk. The chromosome regions specifically examined for sperm mutations in healthy donors were: 7q11.23 (Williams syndrome), 15q11-13 (Prader-Willi syndrome), and 22q11 (22q11 deletion syndrome) and most genetic and epigenetic cases of Williams syndrome, Prader-Willi syndrome and 22q11 deletion syndrome primarily are caused by de novo gene mutations in contrast to being inherited events. Sperm mutations (deletions and duplications) in all three regions were found in the sperm of all the volunteer healthy donors. The frequency of sperm deletions were higher than general population prevalence for Williams Syndrome, Prader-Willi Syndrome and 22q11 Deletion Syndrome suggesting (a) increased fetal loss due to the gene mutation, (b) reduced motility of sperm mutations and/or (c) underestimates of population prevalence for Williams syndrome, Prader-Willi syndrome and 22q11 deletion syndrome. The implication being that all men and women are at risk, however small, for producing a child with a genetic syndrome.
Molina O, Anton E, Vidal F, Blanco J. Hum Genet. Sperm rates of 7q11.23, 15q11q13, 22q11.2 deletions and duplications: a FISH approach. 2011 Jan; 129(1):35-44. Epub Oct 8.
http://www.ncbi.nlm.nih.gov/pubmed/20931230
Yes, I can easily believe the frequency of these de novo mutations is much higher than most imagine. I’m sure there are quality of some of these local genomic regions that are “weak” in some way and lead to more frequent mutations. Fragile X is another prime example, even though it’s not caused (usually) by point mutations but by trinucleotide expansions. That region of DNA is known to be an official “fragile site” in the human genome.