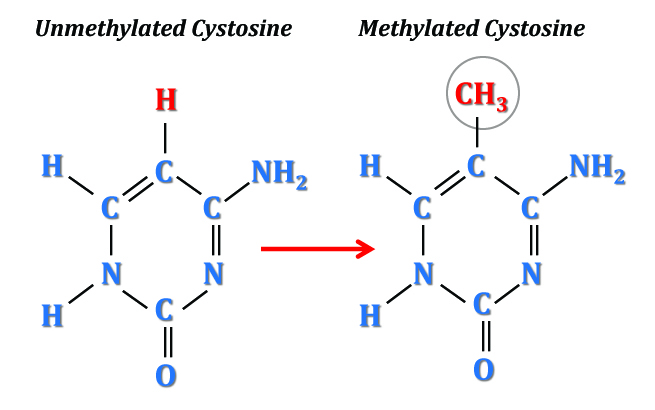
The Methylome is a new toy of genetic science. It adds yet another level of complexity “above genetics”, responsible for affecting patterns of gene expression and, relevant to my line of investigation, perhaps even playing a role in genome stability. But what exactly is it? To put it scientifically, the term refers to the genome-wide methylation patterns seen on the nucleotides, cytosine and adenine. To put it more simply, “methylated” means that a methyl group (CH3) has been added to a DNA molecule. In this case, as illustrated by the image below, the methyl group has replaced a hydrogen (H) atom off of the 5th-position carbon (C).
It’s responsible for aspects of imprinting and X-inactivation, and in general it plays a vitally important role in regulating gene expression by stabilizing DNA structure in a closed (or even sometimes an alternative) conformation [1]. Meanwhile it’s patterns continue to change throughout the lifespan, often times adding more methyl groups to a local site as the years progress.
I won’t profess to be any sort of expert when it comes to the science of DNA methylation. And this blog post isn’t meant to be a lesson from a knowledgeable science teacher to a reader-student. Instead, I am a student along with you and, in finding the whole thing quite fascinating myself, would love to share some of the little treasures I’ve found in a couple articles along the way. So here goes.
I found a letter to Nature by Becker et al. (2011) recently while wandering around Google Scholar seeing what articles would come from the keyword, “methylome”. Even though the paper focuses on cytosine methylation patterns in the plant, Arabidopsis thaliana, it’s relevance to methylation across species is potentially considerable. A. thaliana has been a popular plant amongst geneticists due to its short life cycle and comparably small genome, ranging only approximately 135 million base pairs compared to 3 billion within the human genome.

Image of Arabidopsis thaliana, borrowed from here.
The Becker study sequenced the entire plant’s methylome, comparing rates of epimutation (methylation changes) in the 3rd generation of this plant line to the 31st generation. First off, they found that there were two types of methylation sites: those whose pattern of methylation remained relatively stable across the many generations, termed by the researchers as “non-differentially methylated positions” or N-DMPs, and those which exhibited a certain level of variability across time, called differentially methylated positions (DMPs).
N-DMPs, those which change little over time, on average tended to prefer intergenic (between gene) sites rife with transposable element content (although, in reading the paper and not being a knowledgeable plant geneticist I don’t know whether this refers to active mobile elements or also includes extinct content). From an adaptationist perspective, it is believed that stabler methyl content in these regions prevents transposable elements from jumping around and disturbing the genome, essentially keeping them in “lock down,” something which is particularly important for plants considering they have many more active mobile elements than does our human genome [2]. DMPs, in contrast, were more frequently associated with genes, suggesting that methylation patterns regulating gene expression exhibit much higher rates of epimutation across the generations. In a way this makes sense because gene expression is very dynamic and generally varies across the life span, therefore this part of the methylome needs to be mutable.
Interestingly, though single nucleotide methyl-epimutation occurs much more frequently between generations compared to DNA mutation rates (approximately x 1,000), larger changes in methylated regions appear to epimutate at a similar rate as genetic mutations. In addition, certain sites are especially prone to transgenerational changes in methylation patterns, exhibiting a kind of evolutionary back-and-forth of demethylation/remethylation. Specifically, they found that 32% of DMPs between the 3rd and 31st generations occurred at least twice, and 13% occurred three or more times. If methylation events were random, according to the researchers one would expect less than a 1% reoccurrence.
Now you may be wondering whether methylation patterns might not vary with genetic mutations. Surprisingly, differentially methylated regions (DMR) of the A. thaliana genome tended not to overlap with mutations nor associate with larger genetic lesions, suggesting that epimutation is a divisible process from DNA mutation. In addition, ten times as many DMRs seemed to occur within exons as compared to introns, which the authors suggest may add yet another effector in RNA transcript variation. (During gene expression, the RNA can often be spliced in a number of ways, creating potentially different functional gene products all from the same original gene sequence.)
When the researchers compared the transcriptomes of two randomly selected 31st gen strains against each other as well as two 3rd gen strains, they found that 320 gene products were expressed differently. The two 31st strains exhibited the greatest difference between one another, having been separated from each other by 30 generations, and meanwhile the two 3rd gens exhibited comparably similar transcriptional profiles. In particular, they found that three genes which showed the greatest difference in expression levels between generations and housed particularly conspicuous DMRs, there was a decidedly negative correlation between the level of methylaion and gene expression (i.e., as methylation increases, gene expression goes down). The same relationship, however, did not appear with shorter DMRs and gene expression, suggesting that the extent of methylation can make quite a difference.
Another interesting study by Stadler et al. (2011) reported that DNA-binding sites tend to be methyl-poor regions, indicating that active transcription, as driven by the binding of transcription factors to key sites in the DNA, may discourage methylation of those same sites. Taken together with the information from the study above, it makes sense that genes in contrast to gene-poor regions express greater methyl position variability by nature of their dynamic activities. As gene expression is ever-changing, turning off and turning on again, so too must gene methylation adapt.
But sometimes that adaptation goes awry, as is seen in many cases of cancer. Ehrlich (2002) reviews that both hyper- and hypomethylation of DNA has been associated with cancer development. Of note, however, is that hypermethylation tends to be associated with CpG islands located within genes presumably suppressing transcription of important tumor suppressors; hypomethylation on the other hand associates with repetitive sequences (microsatellites, palindromes) and transposable element content within intronic and intergenic regions. From my own work, it is likely that decreased methylation allows for more frequent open conformation of these regions, increasing torsional strain on the DNA, and impairing stability, ultimately leading to more mutations and other epigenetic disturbances.
As I said, as a scientist I’m still very new to this blossoming line of research. But if one really wants to understand genetics and how DNA behaves through development and over generations, then having a good grasp of current epigenetic theory is undoubtedly a vital part of the geneticist’s arsenal. It’s no longer just an interesting aside we can study, like an addendum to the Book of Genetics, pretending it matters but just not all that much. Every piece matters, especially one so integral to the determination of phenotype. I can imagine that one reason scientists have so hesitantly attempted to integrate these fields is because Genetics is bloody complicated! To add in a whole new Methylome to our conceptual framework can be abominably overwhelming. But for the sake of accuracy and constantly striving to build better theory, we will continue to try. And as so long as we have that little Jiminy Cricket sitting on our shoulders forever reminding us that biology is always more complex than theory no matter how pretty a pink bow that theory is tied up in, our concepts will continue to evolve. Along with our methylome.


Arabidopsis thaliana is at less risk of having high methylome variation over 31 generations than species that produce fewer offspring like us. It is probably important to produce the methylome demonstrated to have worked in the current and related environments. An A. thaliana parent has many opportunities to be assured of achieving that while exploring so much more variation, while we have comparative little. So shouldn’t we have much less methylome variation over the generations, or only limited regions of high variation? Just hypothesizing.
And a very interesting hypothesis. Definitely using not just a different species but a species from an entirely different kingdom than ours certainly raises questions as to whether rates of variation across generations are comparable in the two. Being a professed n00b to patterns of transgenerational methylation, I can’t honestly say I could knowledgeably comment on your hypothesis, except to say it’s a very interesting and certainly a very testable one– excepting of course for the difference in scale, small thaliana genome versus large human one, and also that one would reasonably have fewer generations to observe in humans (4 at most I’d imagine).