This blog post will be the first in a several part series providing an introduction to some of the syndromic forms of autism. I hope readers might find this interesting since we are confronted with so much information in the media about autism but we, myself included, probably have little idea of the numerous documented syndromes that frequently present with autism symptoms as well. Also, however, I hope that lessons can be learned from these syndromes that might act as a guide to understanding what lies at the root of autism, if there is indeed a single generalizable root or common denominator. For me personally, I suspect quite a bit can be gleaned by looking at many of these syndromes together and seeking out commonalities. And because I am unfamiliar with the majority of these syndromes, I’m using this writing opportunity to teach myself. I often find that writing reviews are an excellent way to get ideas percolating. So let’s put on a cuppa. 😀
Timothy Syndrome
Approximately 80% of people diagnosed with Timothy Syndrome (TS) also fit the criteria for autism. It is a potentially deadly condition due to cardiac abnormalities which can lead to fainting and even sudden death. People with TS also tend to have webbing or fusion of the fingers (syndactyly), immune deficiency, hypoglycemia, intellectual disabilities, seizures, and autism [1]. Facial features include a thin upper lip, flat bridge of the nose, and low-set ears.
Most individuals with TS have mutations in exon 8 of the Voltage-dependent L-type Calcium Channel Subunit Alpha-1C (CACNA1C) gene, which causes calcium channels to remain open abnormally long, allowing more calcium into the cells and subsequently activating calcium-dependent pathways. This can lead to changes in cell growth and division, differentiation, cell movement, and general cellular function (physiology). Unfortunately, the condition appears to be autosomal dominant, meaning that if a child inherits even one mutated allele he or she develops the condition. For this reason, most cases are de novo, or arise from new mutations at or near the time of conception, although a small percentage of cases arise from parental mosaicism in which only the parent’s sperm or egg cell carries the mutation.

Example of mild webbing between digits. Image borrowed from here.
Smith-Lemli-Opitz Syndrome
About 70% of people with Smith-Lemli-Opitz Syndrome (SLOS) have a co-occurring autism spectrum condition. Traits of the syndrome include distinctive facial features, microcephaly, intellectual disability and autism, syndactyly (fusion of the fingers/toes) and polydactyly (extra digits), genital abnormalities in males, muscle weakness, and abnormalities of the heart, liver, lungs, and kidneys. Facial features include a tall forehead, flared-looking nostrils, a wide nose bridge, and drooping eyelids.
This syndrome is caused by loss-of-function or reduced-function mutations in the 7-dehydrocholesterol reductase (DHCR7) gene, ultimately promoting decreased stability of the protein. In order for the syndrome to occur, both alleles must be affected, meanwhile carriers of a single mutation do not develop full symptoms. DHCR7 plays a vital role in the synthesis of cholesterol. Without it, cholesterol levels are reduced and its precursors and byproducts build up promoting cellular toxicity. The relative instability of the protein product positively correlates with the level of severity of the syndrome [2].

Image of a child with SLOS. Notice the distinct facial features described above. Image borrowed from here.
CHARGE Syndrome
Autism occurs in about 68% of cases of CHARGE Syndrome, an acronym which stands for: Coloboma, Heart defect, Atresia choanae, Retarded growth and development, Genital abnormality, and Ear abnormality. Most cases present with coloboma, which is a hole within one of the structures of the eye that had failed to close very early in development. Small eyes or microphthalmia are also a common feature, as is narrowing of the nasal cavity or complete blockage from bony or soft tissue obstructions due to failure of the nasal cavity to develop properly. Other symptoms can also include cleft lip and palate, micropenis and undescended testicles in males, delayed puberty, and slow growth beginning in late infancy. Facial features are usually typified by a square shape of the face and notable left-right asymmetry.
Most individuals with CHARGE Syndrome have mutations in the Chromodomain-helicase-DNA-binding Protein 7 (CHD7) gene, which is believed to have some loss of function due to truncation of the protein, affecting its capacity to act as a regulator of gene expression, although its gene targets are not well studied. Most occurrences of the mutation are de novo, although there are occasional examples of an affected parent with CHARGE having children. While autism and intellectual disability are very frequent in this population, there are some individuals whose IQ falls within the average range.

An image of a lovely little girl, Alicja. Read here for more on her story.
Cornelia de Lange Syndrome
A range of approximately 46-67% of people with Cornelia de Lange Syndrome (CdLS) also have autism. Symptoms for CdLS vary widely but often include distinct facial features such as arched eyebrows which join in the middle, a small upturned nose, low-set ears, wide-spaced teeth, and microcephaly. They also often exhibit retarded growth before and after birth and may continue to have a short stature. They also sometimes present with excessive growth of body hair, may develop hearing loss, and have digestive problems.
The most common genetic causes of CdLS are mutations within the Nipped-B-like Protein (NIPBL) gene, although reports of mutations in SMC1A and SMC3 have also been associated with lighter variants of the condition. It is believed that mutations in NIPBL lead to loss-of-function or reduced function in the NIPBL protein, a gene product which plays both a structural role in chromatin and functions in sister chromatid cohesion. As such, it likely plays important roles in mitosis and cell cycle progression.
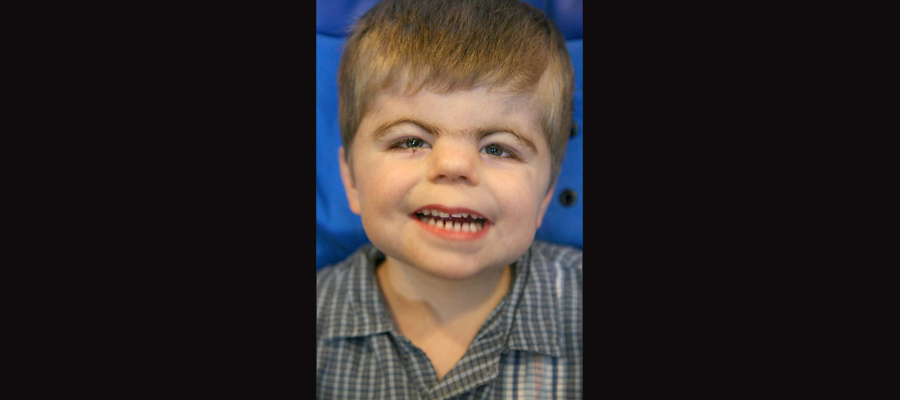
Above is a little boy named Joey who has CdLS. Image borrowed from here.
Lujan-Fryns Syndrome
About 62% of people with Lujan-Fryns Syndrome have autism. Because it is an X-linked condition, it occurs almost exclusively in males. People with LFS are often tall, thin, and have macrocephaly. Facial characteristics frequently include a short philtrum, small chin, and a very prominent nasal root (top of the nose). Weak muscles are another common feature. Seizures as well as abnormalities of the corpus callosum, the white matter fibers which connect the two brain hemispheres, have been reported. Heart defects are also not uncommon.
The mutation responsible for the condition occurs within the Mediator of RNA Polymerase II Transcription Subunit 12 (MED12) gene. Zhou et al. (2012) found that the specific mutation usually associated with the syndrome tends to disrupt an area of the protein which would normally bind cyclin kinase 8 (CDK8). As a complex, this MED12-CDK8 unit normally suppresses GLI3 activity; in the complex’s absence, however, activity of the Sonic Hedgehog (Shh) pathway is upregulated. Shh is very important in organogenesis and as an excreted molecule forms a graded concentration to which cells will react based on the extent of their exposure. This for instance is partly how the dorsal-ventral (back-to-front) patterning is determined within the central nervous system. Shh is released from the ventral (frontal) areas of the brain and spinal cord, meanwhile Bone Morphogenetic Proteins (BMP) are released from the dorsal (posterior) regions. MED12 has also been shown to be a regulator of Wnt pathway activity as well.
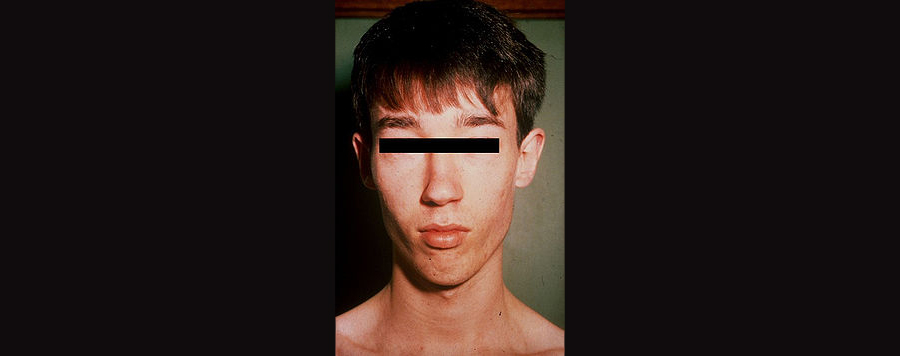
A young man with LFS. Image borrowed from here.
What Do These Conditions Have in Common?
That is the 64 million dollar question. At present it’s hard to even fathom a guess. Aside from autism, all of these syndromes have craniofacial disturbances, a finding which may have links with brain development. At the molecular level, it’s hard to see where these conditions might cross. Here’s a summary:
- Timothy Syndrome is the result of calcium channel overactivity.
- Smith-Lemli-Opitz Syndrome is due to hypocholesterolemia and buildup of toxicity.
- CHARGE Syndrome is due to disruption of gene regulation, although more specific understanding of its cause is still vague.
- Cornelia de Lange Syndrome, whose etiology is also somewhat vague, appears to be due to some sort of disruption in chromatin structure and/or sister chromatid adhesion.
- Lujan-Fryns Syndrome is likely a result of overactivity of the Shh and Wnt pathways in numerous tissues.
Given the likely overactivation of Shh and Wnt pathways in Lujan-Fryns Syndrome, along with its associated macrocephaly, it’s not difficult to see a possible link between LFS and autism, considering the macrocephaly reported more frequently in the idiopathic forms [3]. And perhaps the study of these pathways is a good place to focus in the broader study of autism.
Calcium channel overactivity in SLOS would also feasibly favor neural excitation over inhibition given calcium’s general cellular function as an excitatory ion, and the seizures sometimes present in the condition may bear that hypothesis out. It should be noted that a “hyperexcitable brain” is also an idea not unheard of in the idiopathic autism literature (see Oberman et al., 2010, for example).
The other conditions it’s hard to say. I can’t honestly profess to be an expert in all the effects hypocholesterolemia and toxicity would have on neural function and whether that could lead to overexcitation. I suppose that simply requires more literature searches in the mean time.
The final two conditions, CHARGE Syndrome and Cornelia de Lange Syndrome, I have no idea where to start. The literature on their molecular backgrounds, possible regulatory targets, downstream effects of disrupted chromatin structure or chromatid cohesion… it’s all too vague to make much out of at the moment. But nevertheless, we’ve made a good start with Timothy, Smith-Lemli-Opitz, and Lujan-Fryns Syndromes, so I suppose we can be satisfied with our progress on such a lazy Sunday afternoon. ![]()
Next week, I’ll continue to review several more syndromic forms of autism with high-association, including conditions like Tuberous Sclerosis, Fragile X, and Angelman Syndrome. Stay tuned and keep percolating!

Hi Emily,
the common theme in all these could easily be plasma membrane calcium channel dysregulation and overactivity, at least in those syndromes above where enough is known. Membrane cholesterol (SLOS) is capable of modulating function of voltage gated calcium channels (Timothy syndrome), and the other way round – they occupy a similar molecular location in the membrane http://www.autismcalciumchannelopathy.com/Membrane_Metabolism.html
With regards to LFS: sonic hedgehog signalling regulates calcium spike activity in developing CNS, and again it seems primarily through regulating cellular calcium influx through membrane channels, and “ca spike activity modulates expression of neurotransmitter phenotype [and many other things] in developing neurons … “http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3060219/
There are several other genetic syndromes that often present with autism features http://www.autismcalciumchannelopathy.com/Genetic_Factors.html#
On the Pro side, I could reasonably see calcium playing a role in autism development. On the Con side, calcium is involved either upstream or downstream of almost everything within the cell, so if one goes looking for confirmation of calcium involvement in Such-and-such Pathway, you’ll find it. Which is what makes me hesitant in explaining away Wnt relationship to autism through calcium. I suspect that while a particular Ca2+ channel activity phenotype may predispose towards autism, I imagine there’s a common result which a variety of phenotypes can lead to and which cannot necessarily be explained by a single ion’s activity. I think there’s a process that may be common to all or most of these conditions and there are numerous ways, given the redundancy and crosstalk of molecular pathways, that this process can be instigated or exacerbated. I.e., I’d caution explaining everything with a single Holy Grail such as calcium. –Not to say that it isn’t an excellent line of investigation that could lend much towards knowledge of etiology in general though.
PS: Still mulling over ideas on ERV. Not ignoring your comments/questions. I’m a slow methodical thinker.
Pingback: Syndromic Forms of Autism: Part I | Cortical Chauvinism·
I look forward to the rest of your series. The two most common syndromes associated with high autism risk are Down syndrome and Klinefelter syndrome. The Down syndrome prevelance is 1 in 750 pregnancies. Klinefelter syndrome and the related XYY syndrome occur in males only and the prevelance rates of these two related genetic syndromes are 1 in 500 -1,000. It’s puzzling why Klinefelter Syndrome has been ignored by the autism research community. Bishop’s group in the UK and Ross’s group in the US have found high rates of co-occuring autism with Bishop’s group finding 11% of Klinefelter boys met diagnostic criteria for autism and 19% of boys diagnosed with XYY syndrome meeting diagnostic criteria for autism. Down syndrome, Klinefelter syndrome and XYY syndrome are not inherited
I hope you include these genetically determined syndromes in future posts..
Thanks for that. Yes I agree that it would not be the Holy Grail, and even if it does play a major pathological role there will probably be lots of exceptions. The second important issue of course even if the theory is (mostly) correct would it really open any new treatment directions?
Two things worth mentioning though is Perisico group findings of increased neuronal cytosol calcium levels in autism brain being upstream of mito dysfunction (transferring ASD cytosol to healthy neurons resulted in them soon resembling autism ones in no time – I highly recommend watching http://www.ctl1.com/publicaccess/humanecology/hdru-20091022-eng-ap/# their calcium findings about 2/3 way in). Again not a proof of anything as such but highly ‘interesting’ in this context. Second is reports by parents on some of their children’s ASD symptoms being worsened by calcium supplementation, including downright regressions – children presenting with more severe forms of autism when being given higher doses of supplemental calcium, and bouncing back to baseline after stopping… All anecdotal but would be well worth investigating …
Yes, I agree. There seems to be some good supporting evidence to look further involve Ca2+ involvement in autism.
One thing that this conversation reminded me of, what with some of my research interests in the safety of prenatal ultrasound, is that when cells are exposed to ultrasound (which varies according to intensity, frequency, and duration of exposure), the US wave can create temporary holes within the membrane (i.e., sonoporation), which subsequently allows in a huge influx of Ca2+. This in fact may be its mechanism of action for eliciting neural excitation during transcranial stimulation. Just a thought.