Last week I penned a blog about my current research foci. One project involves transgenic (mutant) balding mice, meanwhile the other is all about autism genomics. Even though these were not meant to be complementary projects, I started thinking about the common ground which they share, realizing how much these hairless mice have taught me about the processes underlying brain development in autism.
So not unlike the Mad Hatter’s “Why is a raven like a writing desk?” riddle, I ask you, “What do bald mice and autism have in common?”

First, some background. My mutant mice overexpress a particular protein called Noggin within portions of the hair follicle. Initially this leads to loss of hair, but as time continues the progenitor pool continues to grow, and eventually this leads to neoplastic and tumorous growths originating from the hair follicle. This happens because the follicular progenitor keratinocytes, rather than differentiating and producing mature keratinocytes, instead remain in an immature state and continue to produce even more progenitors. Here’s a picture of one of my guys below that I showed last week.

What in the world does hair have to do with autism? At first glance, very little. Until you begin to consider that both the central nervous system and the skin are derived from the same germ layer, the ectoderm. What’s more, this neoplastic hair model has more in common with autism than meets the eye. Let’s whittle it down to a simpler model, ignoring the fact that this is a hair follicle, but instead zeroing in on the characteristics of prolonged stem cell immaturity leading to an expansion of the progenitor pool, ultimately leading to neoplasms and tumors as well as dysplasias (underdevelopment) of the mature tissue, in this case the hair shaft.
So, here’s our equation:
increased progenitor pool –> hyperplasia/dysplasia/neoplasia
The autistic brain is very similar to my bald mice, only it shows evidence of dysplasias and hyperplasias more characteristic of the cortex rather than hair. Being a neuropathologist, much of my fiancé’s work has focused on describing the unique characteristics of the postmortem and in vivo autistic brain. Back in 2002, he and his team reported findings of increased density of minicolumns within the neocortex of autistic individuals [1]. Minicolumns are the vertical arrays of cells whose core contains the excitatory pyramidal neurons which most people visualize when they think of the word “neuron”. These columns also include inhibitory interneurons which modulate both the vertical and lateral signals of the pyramidal neurons, modifying crosstalk between minicolumns. The columns of course also include various glial cells, but not as much work has been done on their involvement.
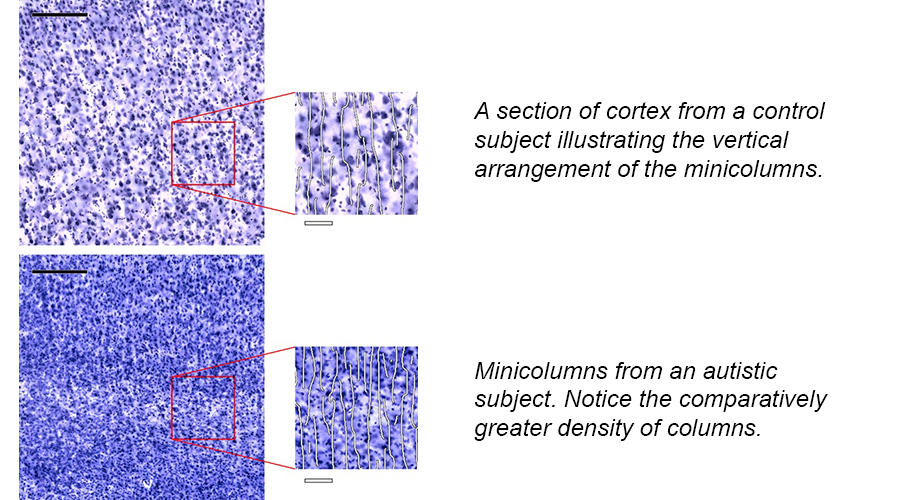
My fiancé’s team went on to simulate whether this increased density of minicolumns simply meant that the columns were closer together or whether this indicated an overall increase in number of columns. Ultimately, their research supports the idea that there are more minicolumns which leads to an increased density of those columns. You might ask how the brain could produce more minicolumns without altering the cell numbers within those columns. Why aren’t there just more cells everywhere? One way to produce more minicolumns is to produce more progenitor cells which are subsequently producing those columns but to not alter the internal “clock” of those cells that tell them when to turn off and stop producing neurons for a single column. So each progenitor is still producing the same numbers of cells for a given column, but there are simply more progenitors producing more minicolumns. This is a hyperplasia of the neocortex, and its mechanisms are very closely linked with those of dysplastic processes.
My partner’s work has also more recently included the study of focal dysplasias in autism [2]. So the autistic brain is very heterogeneous, showing increased growth of the cortex in some areas, and in other locations showing underdevelopment of neocortical tissue. It is quite possible, and even likely, that these underdeveloped areas exhibit an increase in the local progenitor pool which ultimately failed to differentiate and produce enough minicolumns. Evidence for this lies in the occurrence of ecoptic or “misplaced” tissues within the white and gray matter of autistic brains, a finding noted in the large majority of postmortem specimens [3].
In the case of hyperplasia, if you delay maturity of the progenitor pool, this will likely result in an enlargement of the mature tissue, e.g., the cortex. If, on the other hand, if you delay progenitor maturity for even longer, perhaps even permanently, you promote a dysplasia and the tissue fails to develop properly. Both are related by severity, even though one tissue may appear overdeveloped and the other underdeveloped. It’s still likely a similar process that underlies both these occurrences.
So as a recap, in autism we see evidence of prolonged immaturity of the progenitor pool underlying the developing neocortex, as evidenced by increased numbers of minicolumns in some areas, underdeveloped cortex in other areas, and misplaced groups of cells called ectopias within both the gray and white matter, evidence of overproliferation, poor differentiation, and disturbed cell migration.
Now, my mouse model, being a transgenic overexpression model, shows the most extreme version of this type of growth because the protein, Noggin, is constitutively expressed at very high levels within the progenitors of the inner and outer rooth sheaths of the hair follicle. (You generally wouldn’t expect to find an effect this extreme in nature; such Noggin overexpression, without the use of this transgenic system, would kill the embryo very early in development.) But Noggin overexpression initially leads to dysplasia of the hair follicle, in that the progenitors are obviously not differentiating properly and the keratinocytes which supply the formation of the hair shaft ultimately build a very disorganized, malformed hair shaft that often doesn’t even erupt from the skin. Hence, the baldness. Over time, since the Noggin continues to be expressed, the progenitors continue to divide, and divide, and divide, creating more and more progenitors. Until at which point, the hair follicles can become tumorous.
Autism generally isn’t associated with tumors, except in the case of certain syndromic forms associated with gene mutations such as in the tumor suppressors PTEN and TSC2. Why is this so? Well, one could postulate that the endogenous or exogenous effector was weak enough that, though it certainly had a significant result on development, the other endogenous or autonomous cell signals eventually overroad its effects. Another possibility is that whatever this endogenous or exogenous effector was, the progenitor pool was only subjected to it for a limited time, after which the cells were able to resume normal development. This understanding is really important because not only does it give us a very specific time window of vulnerability for autism, it also gives us some ideas as to what effectors could target the progenitors for so limited a time.
You might also be wondering why the autistic brain is so heterogenously affected, showing hyperplasias in some areas and dysplasias in others. The likely answer to this has to do with each cell’s own unique makeup, its location within a given tissue, and its specific neighboring cells. To use my mouse model as an example, all the hair follicles in this hairless model overexpress the protein Noggin. But when you look at a histology cross section of the skin, you can easily see that, even though they all look very disturbed in formation, size, and orientation, each hair follicle is growing in a unique way. Some follicles are gigantic and can even been seen with the naked eye. Others are less deformed and more recognizable as “hair follicles”. A few bizarre ones are growing what I call “pseudo-pseudopods” which are arm-like structures emanating off the follicle. Still others are clearly going through apoptosis and prematurely dying off. Each follicle is unique and reacts to the Noggin overexpression in its own way because each cell has its own unique chemical symphony. There are clearly general trends, but nevertheless you can never fully predict what a single cell, much less a single multicell structure like a hair follicle, will do in the face of such disruption. And this is exactly why there’s such heterogeneity in the autistic brain and across individuals. Even though you may be able to generalize and identify trends in tissue development, ultimately each cell reacts in its own special way. No two cells are alike.
So I hope that after this blog, you can answer the riddle, “What do bald mice and autism have in common?” Unlike the Mad Hatter’s riddle, this does actually have an answer, and one far more important than any raven or writing desk.
