I have to admit that, like most human beings, I can over-focus at times. On this blog, I’ve gone on and on about the embryonic origins of autism and how abnormal proliferation of the progenitor pool underlying structures like the neocortex are at the root of the conditions. I still believe this to be a fundamental finding to most, if not all, of autism’s variants. However, it’s nice to be reminded once in awhile that every theory, no matter how useful, is often too generalized, too black-or-white, and that the truth generally lies somewhere in between. In this case, I am reminded that autism isn’t just about the numbers of cells in the brain that are affected, but that related processes such as dendrite and synapse formation are often targeted too because they all share similar molecular roots.
I attended a lecture last week given by Guoping Feng, an extremely talented researcher currently working at MIT. (I might also add that he seems like an all-around nice guy and had an opportunity for a good chat with him.) Amongst a variety of accomplishments, his more recent work has focused on creating a solid mouse model to study obsessive-compulsive/repetitive behavior. This invariably led to his interest in autism. With his background in molecular genetics, he has focused on the SHANK3 pathway, including SAPAP3, a molecule which binds to and interacts with the SHANK proteins. Both the SHANK2 and SHANK3 genes have strong ties with autism, not only as strong candidate genes in terms of mutations, but also research reported just this month has shown that approximately ~15% of autistic brains in their study sample exhibited hypermethylation of the SHANK3 gene [1].
Research on OCD suggests a disturbed, essentially “overexcited” striatal thalamocortical loop which promotes repetitive compulsive-like behaviors, akin to a record player getting stuck on one track. Dr. Feng was especially interested in SHANK3 because it is the primary SHANK protein that is highly expressed in both the striatum and the neocortex.
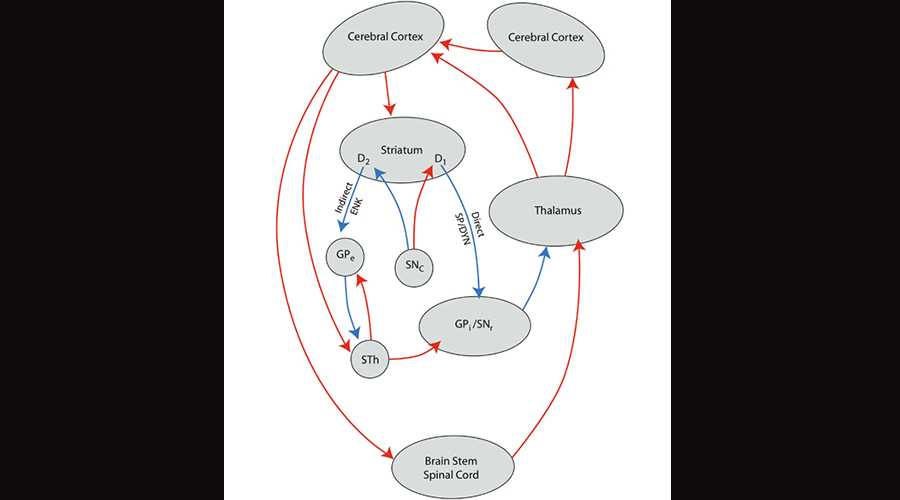
Illustration of the striatal thalamocortical loop, in which the striatum in the limbic system feeds into the thalamus in both a direct and indirect manner, and finally into the neocortex. Image borrowed from here.
Dr. Feng developed two separate mouse models to study repetitive behavior in mice, both a SHANK3 and a SAPAP3 knockout. Mice whose brains do not make these proteins show considerable compulsive grooming behavior, to the point they clean themselves bloody and create considerable facial scarring. In fact, while most mice tend to sleep through the day and are active at night, these null mice spend a surprising amount of their days continuing to clean themselves. Genetic rescue of these mice using a tamoxifen-induced knock-in completely reversed the compulsive behavior after it had started, offering hope that such behaviors may in fact be treatable in a variety of human conditions.
In relation to my own interests, I can currently find no clear evidence that SHANK3 is more than just a synaptic protein but is involved in multiple aspects of cell growth. However, there are a couple lines of evidence to suggest that it may well function in multiple processes:
- Numerous other “synaptic” proteins involved in either cell adhesion or cytoskeletal rearrangements also play roles in proliferation and neuronal differentiation (e.g., CNTNAP2, CASK, etc.).
- SHANK proteins associate with Neuroligins, which in turn bind Neurexins within the synaptic cleft. Knockout studies of both Neuroligin 4 on the X chromosome and Neurexin 1 exhibit disturbances in neuron differentiation, suggesting that they target more than just the cytoskeletal rearrangement of the synapse and neuronal arbors.
So it seems possible, perhaps even probable, that SHANK3 mutations not only affect synaptic development as has gotten a lot of press in the autism research community, but that they play more general roles across the cell in all types of growth. But we’ll have to see about that as more work is done on this fascinating gene, its product, and related pathways.
As I said though, it’s a great reminder that while I don’t think the etiology of autism lies solely at the synapse as is postulated by many current researchers, it isn’t NOT at the synapse either. Instead, it’s probably everywhere; in every aspect of cell growth, from proliferation, to arborization, to synaptogenesis. This is the only conclusion, as I currently see it, that makes the most of the widest range of evidences we have on autism. It explains big brains, it explains increased minicolumns, it explains underdeveloped dysplastic areas of brain, it explains altered gyral patterns, it explains seizures, it explains obsessive-compulsive behaviors, it explains models like the Valproic Acid Rat Model of Autism, it explains the talents and intelligences than can come with the condition.
That’s my prediction at least. We’ll see whether time considers it fruitful or not. In the meantime, I highly recommend to readers that you take a wander around Dr. Guoping Feng’s publications. He’s got some great materials and great ideas. I’m thoroughly looking forward to reading more of his work as it comes out.

Interest in SHANK3 derived originally from the discovery of the Phelin-McDermid Syndrome which features the loss or damage to SHANK3. The question to be asked is where do these mutations come from? Studying consequences but ignoring origins is particularly uninformative. The majoity of cases diagnosed with Phelan-McDermid syndrome occurs as a de novo reproductive error (sperm or egg mutations). Transgenerational inheritance does occur in a minority of cases but when it does the parent(s) are unaffected.
http://ghr.nlm.nih.gov/condition/22q133-deletion-syndrome
I agree completely, Robert. It’s vital to understand when and why these mutations arise. SHANK3 for instance appears to have had a potentially active mutation rate over the last 75 million years presumably, given the considerable differences in amino acid sequence between mouse and man. This suggests there may be elements to this gene underlying its instability, making it a more frequent target in genome destabilization.