“This raises a question: if merely reading a genome differently can change organisms so wildly, why bother rewriting the genome to evolve? How vital, really, are actual changes in the genetic code? Do we even need DNA changes to adapt to new environments? Is the importance of the gene as the driver of evolution being overplayed?”
These are questions David Dobbs recently raised in his article, “Die, Selfish Gene, Die”. I must admit that, though I primarily work with genomics and not epigenomics, my interest in the evolution of mutability was sparked from the idea that epigenetics helps to orchestrate relative DNA stability. So perhaps then it’ll come as little surprise that I commend him for bringing more of the limelight to Dr. Mary Jane West-Eberhard and her work on how phenotype often leads genotype, rather than the other way around. (I do actually have her heavy 800-page tome, Developmental Plasticity and Evolution, sitting on my shelf at home.) And it was some of her work that sparked my imagination, prompting me to wonder by what means phenotype could literally drive genotype, aside from just chance mutation. Perhaps it’s not coincidence then that the act of gene expression increases the risk of mutation, eh?
In any case, I’m not going to delve into a critique of Dobbs’ article, since there’s plenty of scientists who are already both commending and deriding it. I, instead, was struck by the prose above, in which Dobbs questions why, since most gene coding sequences are surprisingly stable across most organisms (as he says, you are after all 80% cow), why do genes still mutate? Do they still need to mutate? Why isn’t this silly tactic of the long-past not given up by now for greater gene stability? Why in the world aren’t we all cows? Or platypuses? Or fish?
The problem is this: If you wish to change gene expression, you either have to alter the molecules which are binding to the genes and altering their expressions OR you can also change segments of the genes which provide a bevvy of different platform options for said binding partners. Often, these numerous platforms reside in promoter, intronic, or even intergenic regions, rather than falling within the coding sequences themselves. Obviously, this latter option necessitates genetic mutation. The bonus here being that, sometimes, unlike binding partners, DNA has a certain level of reliability and therefore reproducibility across the generations.
Let me give an example by using the gene, Glial Fibrillary Acidic Protein, otherwise known as GFAP. This gene’s protein product is a component of the cell’s cytoskeleton, is expressed throughout a variety of tissue types especially within the central nervous system, and is shared by both mouse and human. As may come as little surprise, the human and mouse amino acid sequence similarity is about a 91% match. But humans exhibit some GFAP expression patterns in tissues that the mouse doesn’t share with us, such as within hair follicles. Why might that be?
One possibility is that even though mouse and human share considerable sequence similarity, this gene has different promoters in these two species, leading to different patterns of expression. Specifically, the human promoter lies further downstream of the mouse promoter and is a nonhomologous (different) sequence. Just for the fact that these same genes have two different promoters suggests that they could potentially have different regulatory binding partners, thus leading to different expression patterns. This also means that the transcripts, the basic RNA sequence produced during gene expression, are inherently different, particularly that of the first exons. Even though virtually the same protein sequence is produced from these genes, mouse also produces some extra RNA transcript from exon 1 that is removed prior to protein translation and may provide a function for the mouse that is absent in humans.
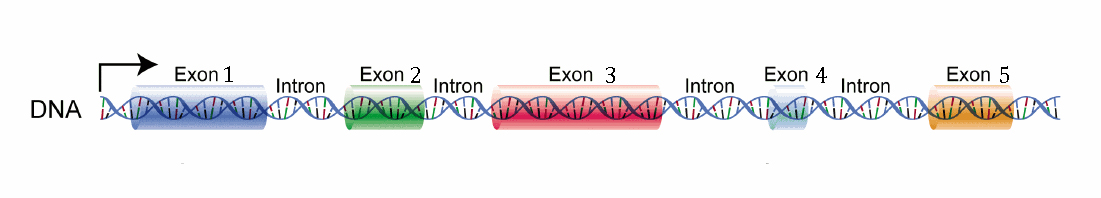
Take the DNA sequence above, a nondescript gene with 5 exons. Exons are the portion of DNA that are eventually translated into protein. The intronic sequences between the exons are not usually translated (although this can vary between different variants of a gene transcript, in which genes often have more than one). In this instance, Exon 1 will end up being our untranslated region (UTR), whereas Exons 2-5 will end up being used for making the protein. During gene transcription, all exons are transcribed, but once that’s finished, the portion of RNA that came from Exon 1 is cut off and intronic sequences are spliced out.
But this brings up another point that Dobbs doesn’t consider: not all gene products are proteins. This includes a lot of RNA products that have physiological functions of their own and play important roles in numerous cellular processes, including gene regulation [e.g. 2]. So even though our proteins may be 80% cow, that doesn’t mean that all our gene products are necessarily 80% cow. There are probably a lot more variations in RNA gene products given the numerous means for splicing and regulation available to them.
Just taking human and mouse again for instance, the Alu mobile elements (small DNA sequences in our genomes which have or once had the ability to make copies of themselves that could move around and reinsert elsewhere) have had a particularly successful run in primate genomes. Meanwhile, they’re all but absent from the mouse. These short mobile elements have been particularly adept at inserting into and becoming part of the exons of various genes in humans. Some of these have become part of the eventual protein coding sequence, meanwhile others are expressed as RNA but are lopped off before translation. Other mobile elements, aside from the Alus, have also generally been more successful in humans than mouse at slipping in and latching onto the exonic machinery [2]. And in fact, insertions of mobile elements in and around genes offer continually shifting means for gene regulation because they often carry regulatory platforms within their own short sequences, e.g., the well know TATA box.
So, hopefully after reading some of these examples, you can start to see why fluctuations in gene expression still often require changes in the genome itself. Mutations aren’t just about targeting the coding sequence but are also about targeting those non-coding sequences within and surrounding the gene that lead to the variability of its regulation. However, this doesn’t speak against West-Eberhard’s idea that phenotype often leads genotype. But keep in mind that the epigenome plays a vitally important role in regulating DNA stability, such that changes in gene expression could very well lead directly to changes in genotype. Truthfully, the idea of “random mutation” never sat well with me. How could anything under Natural Law be random? Chemistry doesn’t behave randomly, nor should biology. The genome has evolved the ability to harness mutability in its favor and, with some wiggle room and plenty of mistakes along the way, to drive it in one direction or another. It’s why you’re not a cow, a platypus, or even a fish.
