I’ve talked about Fragile X Syndrome before and its relationship to autism, albeit briefly. What I didn’t touch on, however, is that Fragile X Syndrome (FXS) isn’t the only condition associated with mutations in the FMR1 gene.
The FMR1 gene lies within a common fragile site, FRAXA, within the X chromosome at the q27.3 locus and produces a protein product, FMRP, that plays important roles in development especially within the brain. The gene houses a CGG trinucleotide repeat in its 5-prime untranslated region (the portion of the exon that is transcribed into RNA but is spliced off before the RNA is sent on to be translated into protein). Most people who fall within the “normal” range have genomes which generally contain anywhere from 5 to 44 CGG repeats; people who have between 45-54 fall within a “gray zone”; meanwhile people who have 54-200 repeats are considered “carriers”; and finally people who have 200+ repeats generally have the fullblown FXS. Ultimately, in FXS the region which contains the 200+ CGG repeats becomes highly methylated, locking down transcription of the gene. Therefore, this condition is one of protein deficiency.
When we talk about the term “carrier”, however, this is actually misleading, because many of these people are not asymptomatic and may even develop their own unique constellation of symptoms, which include neurodegeneration, premature ovarian failure (early menopause), specific learning disabilities or occasionally mental retardation, and autism. Carriers of the premutation, especially males who have between 80-100 CGG repeats, often develop a neurodegenerative condition called Fragile X-associated Tremor/Ataxia Syndrome or FXTAS for short. Usually symptoms begin no earlier than age 50 and primarily target males, although occasionally females are affected too, and according to Hagerman and Hagerman (2006) symptoms include “gait ataxia, progressive intention tremor, parkinsonism, and peripheral neuropathy” (p. 165). In fact, the authors report that scientists and clinicians have begun screening for FXTAS in patients in some adult neurology units who come in for treatment of essential tremor, ataxia, parkinsonism, or multiple system atrophy. They have found that approximately 2-5% of these patients have FX premutations. On autopsy, FXTAS patients often present with inclusions in neurons and astrocytes, with the highest density aggregating in neurons of the hippocampus, which may help pinpoint why FXTAS patients often develop problems with working memory.
What is most fascinating about the FMR1 gene is that, depending on the number of repeats, fullblown syndrome versus premutation, different things can happen to the gene product. In the case of FXS, increased trinucleotide repeats promote hypermethylation and prevent the gene from being transcribed. In the case of conditions like FXTAS and premature ovarian failure, these instead appear to be due to increased transcription of the FMR1 gene. In other words, these conditions are the result of RNA toxicity and gain-of-function as opposed to the protein deficiency that typifies FXS. Although it’s not precisely understood, a moderate expansion of CGG repeats in the 5′ untranslated region of FMR1 promotes increased transcription and more gene product. In the case of FXTAS whose risk increases with age, the cell might find it difficult to dispose of all the extra gene product which eventually builds up and causes problems in the machinery of the cell. This is actually quite similar to the well known amyloid plaques that occur in the brains of people with Alzheimer’s Disease. Often, the RNA or peptide can bind unnaturally to itself or with other proteins, thereby preventing it from crossing cell membranes. This process is called “fibrilization”. In order to be dealt with, the RNA or proteins must be reshaped into their proper forms by players such as heat shock proteins, although this is not always successful. This is a common occurrence with gene products which are overexpressed.
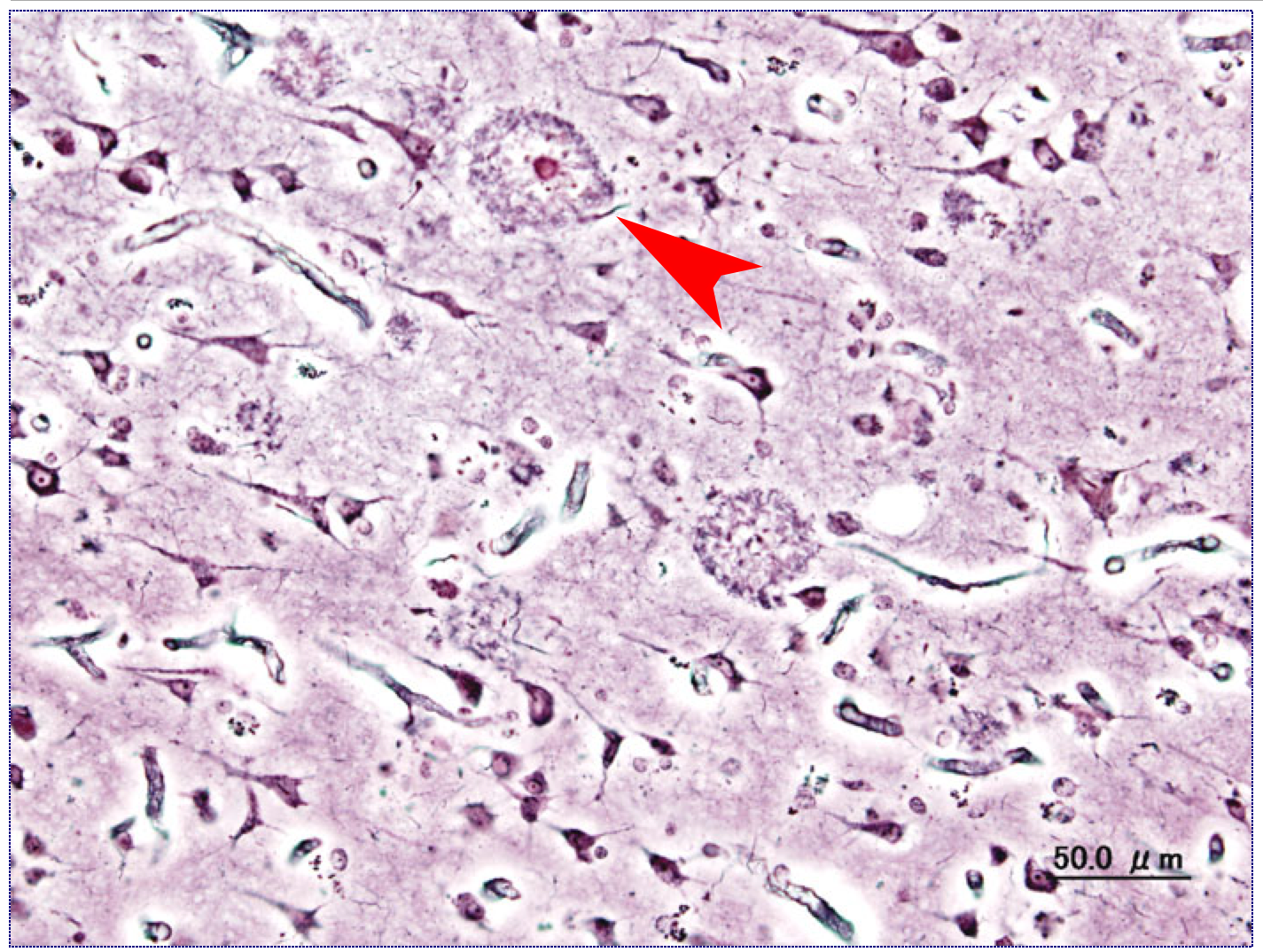
An example of an amyloid plaque commonly seen in Alzheimer’s Disease. In this instance, the central body is the beta-amyloid aggregation while the surround is the plaque itself.
Scientists believe that, while symptoms of FXTAS and premature ovarian failure are due to RNA gain-of-function of the FMR1 gene, the fullblown behavioral syndrome as well as the lighter variants which are sometimes seen within the premutation range are the result of FMRP deficiency. This is borne out by the fact that behavioral symptomology and its severity shows a direct inverted relationship with FMRP levels across individuals. In addition, it is believed that some of the physical features associated with the condition and premutation carriers, such as elongated faces, large ears, and hyperflexible joints, are also due to FMRP deficiency.
As you can see, genetics are complicated. On the one hand, increasing CGG repeats in the FMR1 gene, especially between 80-100 repeats, increases gene transcription somehow and produces conditions like FXTAS and premature menopause. On the other hand, go a little higher and the same gene sequence is vulnerable to hypermethylation and transcriptional lockdown, producing the behavioral and physical symptoms described above. So it’s not as simple as scientists and clinicians once thought.
If you’re interested in human conditions due to these kinds of simple genetic instabilities, I highly recommend the text, Genetic Instabilities: Neurological Diseases, edited by Wells and Ashizawa. It’s a great book, not weighed down by huge amounts of technical data, and is a good way to learn about the behavior of repetitive sequences in general.


Emily, I found this article especially interesting. I’ve been suspicious that my hemifacial microsomia (Goldenhars) and Asperger’s might have been the result of a a threshold accumulation of mutations (or, perhaps as you have determined, repeats). As a female I wasn’t thinking in terms of Fragile X, but you’ve got me wondering. No one else in my family has hemifacial microsomia, but my father definitely had Aspie traits. (There’s one of my maternal aunts about whom I sometimes wonder, but she’s long gone, had some profound depressive issues that might confuse the situation, and my cousins might take it badly if I approach them about the possibility.) I had been reflecting on some of the other known neurological and neural tube mutations in my father’s family and how they might fit into the picture. My father had a very obvious case of essential tremors ands one of his sisters has it badly as well. I show no signs, but I haven’t hit 50 yet. You mentioned in passing that essential tremors might be connected with FX premutations. This article seems to confirm the link and has some interesting things to say about possible cognitive comorbidities, though social phobias rather than ASD are mentioned: http://www.medscape.org/viewarticle/740330_5 There also may be a gene in the family for spina bifida occulta. The same aunt who has the essential tremors has that and passed it on to her progeny. I’m not aware of anyone else with this, but it’s possible there may be some carriers who were never caught through back x-rays or just never mentioned it, since it’s asymptomatic. I wish I had the opportunity to have some genetic testing. Date: Mon, 3 Feb 2014 00:25:04 +0000 To: pamelasage3365@outlook.com
It definitely sounds like it could be some sort of genetic syndrome you’re describing, especially like the related case of FXS and FXTAS and repeat instability. If you can get genetic testing, it sounds like it could be useful to you. Although I could understand it may be a challenge in getting the testing in the first place. You’ve probably already done this since you seem to have done a lot of the leg work already, but try talking to your GP about getting a referral. I don’t honestly know whether you’re insurance would approve that (if you’re in the US), but it’s worth a shot if you haven’t already tried to go that route. Getting your condition diagnosed could be important for future treatment plans for you. Good luck and let me know how it goes, Pamela. I wish I could be of more help to you but unfortunately the practical side of medicine isn’t my specialty.