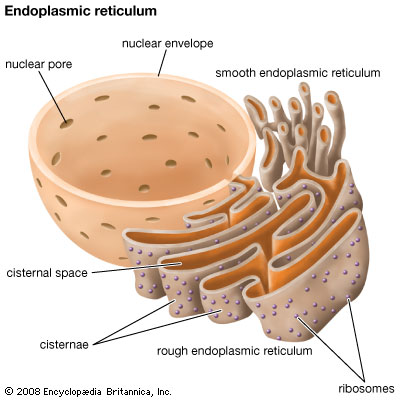
A few weeks ago I wrote about cell stress and its relationship with epilepsy. I reviewed how the accumulation of misfolded proteins within the endoplasmic reticulum (ER) can lead to an Unfolded Protein Response (UPR), which, if chronic, can lead to cell damage and even cell death.
This week, I’m talking about autism.
When we talk about “neurodegeneration”, probably the first thing that comes to mind is something like Alzheimer’s or perhaps even Huntington’s Disease. In these conditions, it’s obvious that there’s a progressive loss of cognitive function and we know that this is accompanied by neuron loss as well. Autism, on the other hand, while it may experience some setbacks in the form of regression, typically follows the opposite progression: it improves over time. So at first glance, one would think that end-of-life neurodegenerative diseases have little in common with a neurodevelopmental condition like autism. However, there may in fact be more there than meets the proverbial eye.
For the last decade, the scientific community, and particularly the geneticists, have been enraptured by potential synaptic involvement in the autism phenotype. There have been a number of candidate risk genes whose gene products function at the synapse (although they may also function elsewhere in the cell), and because of this, enthusiastic scientists have had a tendency to cherry-pick the genetic data to focus on the synapse in autism. (At one point I gave them a greater benefit of the doubt, however my recent work in genetics indicates that there are a comparatively small number of genes in autism that are involved strictly at the synapse and that risk genes tend to affect a broader range of neural development than typically acknowledged. So, um yeah, definitely guilty of some serious cherry-picking.)
In the study of neurodegeneration, it is well-acknowledged that not only may a particular genetic loss- or gain-of-function be responsible for a given condition, but the mutation may lead to a change in the shape of the gene product when one is produced. (This is especially more likely when small mutations such as point mutations occur, rather than a large deletion encompassing a gene.) In these instances, a change in shape can lead to stalled protein processing within the lumen of the ER, causing an accumulation of that gene product. Not only does this mean that the cell is likely haploinsufficient for that product (it only produces half as much as a normal cell), which may or may not impair cell function, but the build-up of protein within the ER can lead to cell stress, triggering the UPR, and can lead to altered physiology and even cell death if chronic or severe enough.
Interestingly, there are a number of studies in autism that suggest this may also be the case, although strangely they have received little press within the scientific community. Back in 2010, Fujita et al. reported on several cases of point mutations within the Cell Adhesion Molecule-1 (CADM1) gene. They found that function may have been only modestly reduced (~20%), suggesting a minor loss-of-function. However, they also noted that the mutated Cadm1 protein exhibited abnormal intracellular accumulation. When they investigated its specific intracellular location, they found that it was abnormally accumulating within the ER. It was also colocalized with a molecule known as beclin, which tends to localize within the ER under ER stress. They concluded that the mutated protein led to its abnormal accumulation within the ER and subsequent activation of an ER stress response, i.e., UPR.
What I find particularly interesting is that, even though the team’s experiments suggested there was only a minor loss-of-function in terms of Cadm1 activity, neurons transfected with the mutant protein exhibited short dendrites or failed to develop dendrites at all. So either a mild insufficiency of Cadm1 is unusually detrimental to dendrite development OR the phenotype is complicated and exaggerated by the ER stress response, the latter of which makes more sense given the UPR’s involvement in regulation of various membrane receptors, such as glutamate and GABA receptors, as well as protein translation and cell growth in general.
Interestingly, several studies have reported similar findings on Neuroligin-3 and Neuroligin-4, indicating that these point mutations likewise lead to the accumulation of their protein products in the ER. Though they’re currently few in number, these studies highlight the fact that ER stress may be an integral part of some individuals’ phenotypes, and may not only help us understand the cause but may also offer some means for amelioration of symptoms.
It also may lead one to wonder how much the loss- or gain-of-function of the actual protein product itself is involved in the individual’s condition or whether the ER stress may itself be a bigger driver of autism risk.
Some evidence suggests that could be the case (although when is biology ever so simple?). For instance, it has been suggested that the UPR can affect and may play important roles in neuronal differentiation, a stage of development I’ve considered far more integral to autism risk [1, 2]. In addition, Momoi et al. (2010) have already proposed that
“[ER stress] arising from these mutations causes a trafficking disorder of synaptic receptors, such as [GABA B-receptors], and leads to their impaired synaptic function and signal transduction. . . . [We] propose a hypothesis that ASD pathogenesis is linked not only to loss-of-function but also to gain-of-function, with an ER stress response to unfolded proteins under the influence of epigenetic factors” (p. 13).
As an addendum to the above statement, I would propose that ER stress affects not only synaptogenesis but likely all stages of neuronal development, as we have been finding.
Again, it is shocking that this work by Fujita and colleagues has not received more attention by mainstream autism science. And yet this area probably holds the greatest promise for treatment intervention. Already, there are a number of medications on the market used to treat neurodegenerative diseases, some as simple as over-the-counter aspirin. Why then has this work been ignored, especially considering the platforms from which the evidence has been shouted, like Nature and The Journal of Neuroscience? Why has this grabbed so few scientists’ attentions? The implications are astounding for genetics research.
Perhaps scientists just don’t get how astounding.

Dear Emily (hi and ola to Michael too for his blog Cortical Chauvanism (oops spelling) as I follow and enjoy both 🙂 ), You said you were going to blog about autism and then started by discussing neurodegenerative conditions (Alzheimer’s and Huntingdon’s Disease aka H’s chorea) . . . I’ve only skim-read so far but I spotted, “There have been a number of candidate risk genes . . . ” Hey, Emily, can I check with you, are you referring to genes that are associated with a higher “RISK” of autism.
This negative type of language is so widespread in the scientific language, mindset, prejudice etc that it gives me a “heart-sink” reaction whenever I read it. Some of the charities (e.g. the dreaded and so often despicable “Autism Speaks”) even promote a negative attitude to autism, because they see it as the best way to raise funds by eliciting sympathy for what they regard as those poor families whose lives are negatively impacted by having an autistic child . . . All of the negative language so frequently used (e.g. “risk” “problems” “deficits”) feeds into prejudice against the autistic community. What about the positive aspects of neurodivergent development, Emily? When is the scientific community going to embrace a truly inclusive approach to autistic people as “different” from the majority of the population (i.e. the “neurotypically developed” 99% or so) and yet still valued members of our human community? Can you help by being more mindful of the language used within your two blogs at least? I am (truly fortunate and blessed to be) a wife, a mother of 3 sons, and a full-time unwaged volunteer as an Independent Autistic Advocate (“ASC adults to . . .” socialise / collaborate / educate / (focus on solutions); I am a graduate of Clare College, Cambridge (MA General Medical Sciences, with elective studies in “Medicine, Ethics and the Law” and in “History of Medicine” (2i) (“boatie” 2ii overall – oarswoman and senior C sculler – though only 116 lb, I won Blondie Colours (half blue) as reserve for CUWBC – 1982), also of the University of London (MB BS, 1985), MRCPath (1994), FRCPath (1999), PG Dip (composition) with distinction from the Royal Northern College of Music (including jazz, musical direction and Dalcroze Eurhythmics, 2008), Grad Dip Ed (Primary) (2011) . . . . who was only identified as being autistic (Asperger’s Syndrome) in August 2011 (diagnosis confirmed 16/02/12) at the age of 50. “I didn’t get where I am today without . . . being autistic”. I have had a 20 year career as an NHS hospital doctor, almost entirely in Cellular Pathology. My autistic personality includes traits that are enormously valuable to society (attention to detail, perseveration, systematisation, perfectionism – well that’s the kind of person who we’d want to have reporting on our FNAs, biopsies, resection specimens and autopsies, wouldn’t you say??? . . . ) My tendency to literal thinking, coupled with my fascination with metaphorical alternative interpretations, has only added to my love of poetry, crosswords, ability to “think outside of the box” and general creativity (appreciation of visual arts, graphic design, animation too). My 30 year “career” as a mental health service user (atypical depression with anxiety, low self esteem, low confidence, recurrent suicidal ideation and, more recently, a tendency to “switch” to a hypomanic type presentation in response to antidepressants, which I term “Iatrogenic Scatterbrain Toxicy” 😉 and also PTSD since 1987, plus worsening post-traumatic-stress-disorder due to the drip-drip-drip torture of being so often misunderstood and denied reasonable adjustments even after declaring all of the above – including instances of what I regard as psychological abuse by professionals who don’t understand autism even though they think they do but don’t even do autism awareness training . . . ) is understandable due to the strains and stresses of living with a “hidden disability” (i.e. autistic minority in a predominantly neurotypically dominated society); the positive aspects of all of that lived-with experience include profound insights, an awareness of group dynamics and interpersonal relationships, self-help cognitive therapy since 1988 (David Burns – Feeling Good – the new mood therapy – a life saver), Levels 2 and 3 certification in Counselling Theory and Practice and, following on from my formal diagnosis as having an Autism Spectrum Condition (btss.org.uk), having benefited from “Solution Focused Brief Therapy” with Vicky Bliss (btss.org.uk . . . buy the book . . . she’s transformed my life from problem-orientated to my Focus on Solutions YAY). In Britain, as well as the Equality Act (2010, currently under review by a Select Committee of the House of Lords, deadline for submissions 11 Sept but may be extended on request as they have done so for me 🙂 . . .) we have an Autism Act 2009 (as well as an Equality Act 2010) Sincerely, Jo, aka NumberNine (aspievillage.co.uk), Joanna Treasure (Dr / Ms), “Peace and light” to all and “May your God go with you”.
Hi, Joanna. I also have a diagnosis of Asperger’s (2007) and, while I’ve moved away from the community nowadays, I spent much of my time very involved with the online autistic community since 2004. I owned my own autism forum (Gestalt) for some time and was also moderator and have been long-time member on WrongPlanet. I also keep in touch with its owner, Alex Plank, and usually see him at IMFAR. I simply give this background to say that I am very familiar with the ND movement and my first major experiences of autism were centered around it. I definitely support the concept that all people are unique and should be treated as such, with love, compassion, support, and understanding, rather than pity and sorrow.
That being said, unfortunately there are certain terms used in the professional genetics literature that communicate particular ideas. While I myself disagree both personally and theoretically with a pathology paradigm, being a professional in this realm I find it almost impossible to communicate my ideas without subsuming much of the terminology. For my own part, I try very hard to avoid the term “disorder” as it adds a layer of judgment to a person’s condition which is unnecessary, not wholly accurate, and potentially detrimental to the person’s sense of self-worth.
From the scientific point of view, however, there are specific genes and specific mutations that increase the probability of developing autism. Much of the risk genes identified to date, however, often only give a snapshot of individuals who tend to be much more severely affected than those with HFA or Asperger’s. Many of them would now be considered “genetic syndromes” that are accompanied by other features. Some of them, though rare, are extremely severe and can lead to severe intellectual disability with accompanying autistic features. (Some people argue that true autism is solely “idiopathic” and that genetic syndromes are different altogether. However, the data do not support that and instead suggest that there are MANY autisms, ranging from low-to-high functioning. Autism is more likely a common anatomy and physiology that many different genetic and environmental background can funnel into.)
In short, the genetic susceptibility is a very real and measurable thing I promise you, though it likely gets much more complicated in the higher-functioning ranges because those genes are likely less penetrant for the phenotype, may involve a number of different genes (polygenic), and probably also involves a certain level of environmental influence.
However, I do understand where you’re coming from, as I have been there myself. But thank you for the reminder. It’s very easy to become too inculcated in a particular paradigm and forget there are other valuable viewpoints out there.
As an aside, I encourage you to read the entirety of the article, as there are distinctive links made between neurodegeneration and autism that might prove interesting.
Hi Dr. Casanova,
I love your analogies and your recent tale of homework, hookey and chaperones. The analogies help when trying to begin to understand some of the complicated subjects you take up on your blog. One of my simplistic imaginings of point mutations is that they might produce slightly deformed proteins that could conceivably function at a reduced level. Does this sometimes happen? If so, does it mean that a protein that doesn’t fold properly, despite chaperone help, can sometimes escape the ERAD degradation process and end up as an inefficient worker at its intended job? Or, on the other hand, are proteins things that aren’t expected to function at all if not built “just right”?
Is the idea more like a point mutation is going to require the ER to do corrective folding on the way to making each proper protein, risking triggering the UPR if enough misfolded proteins accumulate?
Mostly, it’s so interesting to notice that miscoding for a wide variety of proteins can set off common outcomes due to “manufacturing stress.”
Hi, Erica. Most typically if a protein doesn’t fold properly, it tends to be shunted over to ERAD, unless the ER is already overwhelmed by attempting to unfold other proteins. Then I believe a few sneaky ones can get through. This aspect I haven’t studied so well, so I’m as new to it as yourself. Although suffice it to say that a protein can actually fold properly so as to escape detection in the ER but not function properly, so you may still see those effects out in the cell. There are also a few mutation types, especially in the case of small repeat expansions, that lead to an expansion of certain amino acid strings (poly-glutamine, called poly-Q chains) that can cause the deformed proteins to aggregate and stick together. These would be acting somewhat as “prions”, similar to how a foreign or native protein, such as in mad cow disease, can trigger other proteins to misfold by binding to them, thus starting a long chain reaction. I’m not saying that this reaction is necessarily occurring in autism, because typically it’s association with very distinctive neurodegeneration. However, I thought the concept worth sharing.
Hi again Dr. Casanova,
You’ve been of so much help! Hopefully, I can now in turn help, at least to the extent of answering some of my own questions, as to reduce total questions asked. : )
Along those lines, I found a series of articles (one of which contains graphic below) that mostly took care of inquiries from my earlier comment. Thank you for guiding attention to all this!