Ever since synaptic gene mutations were discovered in autism, the scientific world has been obsessed [1, 2]. Interestingly, however, these synapse-specific genes make up only a small minority of autism risk genes, in spite of the considerable amount of scientific attention they garner. In our recent publication, we found that synapse-related genes make up only about 10-13% of extremely high risk autism genes, meanwhile the bulk of gene products function within the nucleus as gene expression regulators. Obviously, the regulators can and do affect the function of synapses, but their consequences are cell- and tissue-wide, making the situation far more complicated than just synaptic dysfunction.
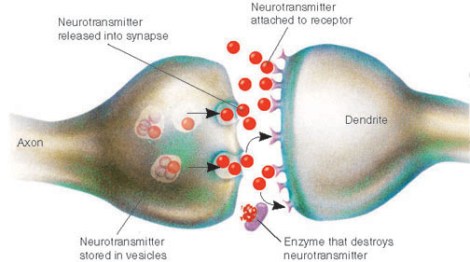
While synaptic dysfunction in the mature neuron is undoubtedly a vital area of study, unfortunately scientists have almost entirely ignored the roles of these synaptic proteins in early development of the prenatal brain. For instance, did you know that even back in the 1990s, some scientists realized that neurexins, a family of synaptic cell adhesion proteins associated with autism risk, are expressed in newborn neurons in mouse brain as early as embryonic day 10? [3] This means that these synaptic proteins are likely playing roles in newborn neurons long before they ever reach their final destinations, sprout neurites, and develop mature synapses. (One possibility is that these early-expressed synaptic proteins are helping to field incoming excitatory signals from cells such as the GABAergic interneurons that drive the maturation of newborn neurons as well as some progenitors. A loss of these signals would be catastrophic to differentiation and survival.)
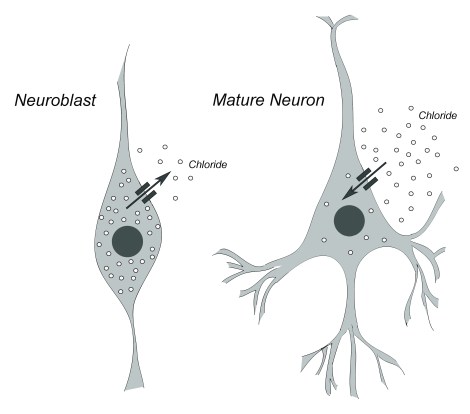
In newborn neurons, GABA has an excitatory effect, while in mature neurons it serves an inhibitory function. This switch is controlled by the relative amounts of the negatively-charged ion, chloride, both inside and outside the cell.
Evidence from mutations in the CNTNAP2 gene, which is part of the neurexin family, also suggest its importance in neuron development before and during neuronal migration. It’s been found that mice with a double knockout of the gene exhibit migrational abnormalities in the form of ectopias (misplaced cells), a finding that echoes human cases of Cortical Dysplasia-Focal Epilepsy (CDFE) syndrome that are linked with CNTNAP2 mutation [4, 5].
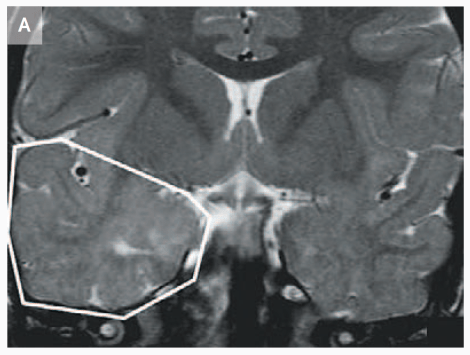
MRI that shows increased volume in the temporal lobe of a 3 year-old child with CDFE syndrome (Strauss et al., 2006).
Interestingly, and for reasons not currently understood, mutations in various synaptic genes, such as NLGN4, CNTNAP2, and CADM1, have all shown evidence that suggests not only do these mutations promote functional losses at the cell surface, but specific mutation types in these genes produce a protein product that has a tendency to accumulate within the endoplasmic reticulum (ER), which is the protein-producing factory for the cell [6, 7, 8]. This accumulation suggests that these proteins are misfolded due to the mutation and are aggregating in the ER, which is probably triggering a cell stress response to deal with the backlog. Such a cell stress response has the capacity to disturb early brain development severely, not only altering differentiation but, if severe enough, promoting cell death [9].
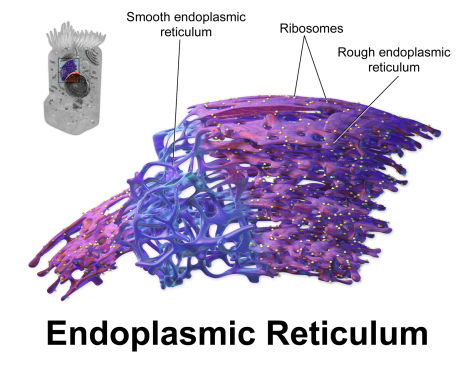
Why has this last finding so far only been reported in association with synaptic gene products? Well, it could be coincidence, which is entirely possible and this should be addressed. However, another thought that springs to mind is the fact that synaptic proteins (and the genes they’re derived from) tend to be very long, which suggests a certain level of functional complexity. It’s possible that long, complex proteins may be at higher risk for misfolding and “gumming up the works” so to speak– although that is only a vague, though testable, hypothesis at the moment.
All of this information does suggest, however, that synaptic dysfunction during development is far more complex than looking at the structure and function of adult synapses. If we want to understand the larger picture, we also need to understand the relevance of these mutations to early development of the brain, in both structure and function. And we must also determine whether the phenotypes we’re looking at in human cases are simply due to haploinsufficiency of the synaptic protein or whether there’s another layer of complexity (e.g., cellular stress) that is also playing a vital role in autism risk.

Hi Emily
This is a very interesting post on the neural structure of the brain and how this relates to autism – from a personal perspective there could well have been genetic factors (in terms of brain development) – many of my additional disabilities in context were caused by a placental abruption (lack of oxygen/hypoxia/brain damage) – I was also premature and the potential for a pre-natal stroke (causing addition damage) cannot be ruled out also.
That could mean that the neurons “dying” rather then being “grown with mutation” – my speech and language developmental was poor and certainly has an aphasia both receptive and expressive and would have been in the sub-group for a time non-fluent (patterned language, echolalia, pro-noun reversal, pre-literal, creating my own language before “interpretive language” relying on gesture, pantomime and “over the top” vocals (speech).
Bilateral damage to my occipital lobes was also present my mum thought was deaf and blind – Olga Bogdashina talks about “blindisms” and “deafisms” that certainly was the case for me with severe simultagnosia (seeing things in bits), semantic agnosia (my body such as mouth, tongue, lips, feet, hands to gain a reality) and not being able to “internalise” language with picture also hampered my language development also. These aspects still effect me (I cannot visualise so I do semantic associative behaviours such as having everything on display and in placement so it has a context), receptive language means my ability to “hear” becomes “distorted so even the point of the literal of what the person is saying is “missed” so tone, gesture, placement and lists are helpful indeed.
As I know both yourself and your Father are very perceptive and compassionate people on the field of developmental disabilities – could neurons in my case have “found another pathway” as time went by which also connects with the environment I was in? 🙂
Kindest regards
Paul Isaacs
Prematurity can definitely alter the timing of brain maturation, which may be why prematurity is a risk factor for autism and other neurodevelopmental conditions. Your case is probably more complex than that but that’s just off the top of my head.
As an aside, Manny is my husband. Although given our age difference, the mistake is understandable. 😉
Hi Emily
Oh Emily my sincerest apologies to yourself and Manuel thank you for being understanding. 🙂
Thank you for your input it certainly seems that brain injury altered my development as well as being premature I was about a month premature so there was a level of underdevelopment so I also a level of hemiplegia left hemisphere disconnect so that means I have problems processing my right side, motor coordination. visual, spatial issues persist but over time things have improved with swimming, bike riding and walking over the years. 🙂
Kindest regards
Paul