Over the last year and a half, I’ve been studying rare forms of intellectual disability (ID) that have single-gene (monogenic) origins using various bioinformatics approaches. My primary interest was to try to determine what relationship lay between ID, autism, and epilepsy and to figure out when those conditions tended to overlap. Did they usually overlap in specific genetic conditions and not so in others? If so, are we able to see functional trends in the genes that underlie these different conditions?
As reported in our latest publication published in Molecular Autism a few days ago, we divided our conditions of interest into several groups according to their comorbidities with autism and epilepsy as well as the frequency of those comorbidities. Ultimately, we ended up with five basic groups:
- ID with high autism comorbidity rates (20+%) with/without epilepsy
- ID with variable autism comorbidity rates (>20%) with/without epilepsy
- ID with high rates of epilepsy (20+%) but no autism
- ID with variable rates of epilepsy (>20%) but no autism
- ID without either autism or epilepsy
We specifically collected together IDs that had single-gene origins, as opposed to large deletion or duplication syndromes, such as Williams Syndrome that involves deletion of the long arm of chromosome 7 and therefore multiple potential gene targets. This allowed us to whittle down the comorbidity relationships roughly to a single gene, whose function then can be analyzed. (Disclaimer: When we say “single-gene” this doesn’t mean that the gene mutation is the only factor affecting the presentation of the condition. We just mean that we know the gene mutation is the major factor.)
The major finding of our study involved those genes associated with ID with high rates of autism comorbidity (HCA). Compared to all other groups, we found that the HCA gene group was heavily involved in the regulation of gene expression and that their gene products tended to function within the nucleus and are linked with structural development of different tissue systems. In short, they were nuclear epigenetic regulators, such as transcription factors/repressors, methylators, histone modifiers, and ubiquitinases. You can see in the bar graph below that, compared to all our other comorbidity groups, the HCA group had extremely high rates of these regulators.
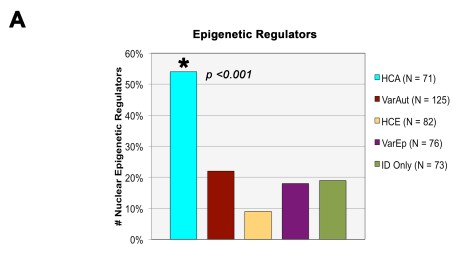
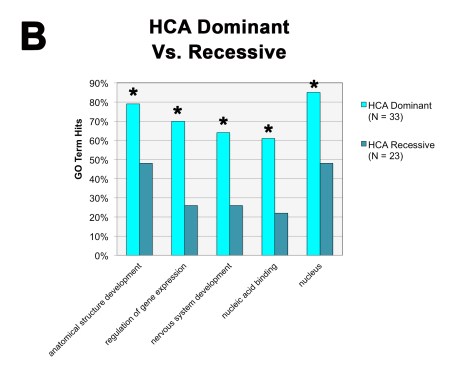
We also found that, when we divided the HCA gene group according to the inheritance patterns of the IDs themselves (dominant vs. recessive), the dominant HCA gene group was primarily responsible for the bulk of the general HCA findings: they were located within the nucleus and directly regulated gene expression. The recessive group on the other hand didn’t show any of these same trends.
So not only are these types of nuclear epigenetic regulators highly penetrant for the autism phenotype, they are highly penetrant (i.e., dominant) in general and haploinsufficiency can obviously lead to some pretty detrimental effects.
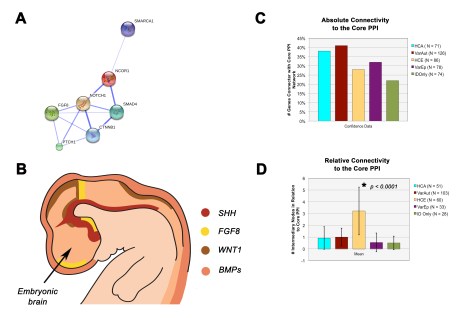
Finally, we found that when we performed a protein-protein interaction (PPI) network analysis, both the HCA and variable autism groups were more strongly and tightly connected to a molecular patterning network that is foundational to the organizing centers of the embryonic brain (see panels A and B in figure below). This suggests that in these monogenic forms of ID with autism, basic patterning of the central nervous system may be disturbed. Further work is needed to address this possibility, which we hope to be following up using specific mouse models. It will also be interesting to determine whether this is something that may be specific to these genetic syndromes of autism or whether it could be something that the broader spectrum also shares in common.
On an interesting side note, close to half of the genes we studied in the HCA group are not currently contained in the SFARI Gene Database, are included but aren’t scored, or are scored as a “6” which means that a link to autism is unlikely. This suggests that the SFARI database may be lacking some vital information in the study of autism. In addition, 69% of the genes that we studied that were ranked within the database and are not considered “syndromic” do actually exhibit additional features (e.g., facial malformations) that would indicate they are definitely syndromes in the classic sense of the word. (Note: “Syndrome” can mean any consistently occurring constellation of symptoms and so autism would be considered a syndrome. However, when studying genetic conditions, “syndrome” typically refers to a constellation of symptoms that occur across different tissue types, such as the brain, the skeletal system, and the heart. Some genetic forms of ID have other physical symptoms and are referred to as “syndromic”, meanwhile other types of ID only appear to affect the brain and don’t involve disturbances to other systems or if they do it doesn’t occur in a consistent fashion enough to be considered a “syndrome”. And therefore, in the study of rare genetic conditions, they are called either “syndromic” or “nonsyndromic”.)
As a recap:
- Genes with high penetrance for ID with autism tend to produce proteins that function within the nucleus
- These proteins typically regulate gene expression
- They are often involved in the structural development of different tissue systems, including the brain
- Highly deleterious mutations in these genes are usually inherited in a dominant fashion
We are continuing our study of these various ID groups, but have moved beyond the level of the gene and are now completing a study looking at variations in phenotype associated with the different ID groups. Though I can’t currently share our findings, I can say that we’ve definitely found some significant trends when comparing ID with high rates of autism to other forms of ID. More to come on that in future…
