
My partner has adored chess ever since he was a child.While I’m no aficiando like he is, there’s something easy to romanticize about the game. The sheer artistry of the pieces themselves, the beautiful organization of the alternating black and white squares, and the intellectual skill a game such as this demands. It’s easy to see how its popularity has stood the test of time and maintains an ardent group of enthusiastic devotees.
In order to be a successful chess player, one must be capable of thinking numerous moves ahead and entertaining a variety of possible alternatives. This is the primary reason the majority of people are not good at chess because, though humans are arguably the smartest animals on earth, our predictive and multitasking capacities are still, well– shall we say “less than desirable”? And I certainly don’t exclude myself from this group when it comes to chess. I’m afraid I’m more a collector than a player.
Biology, on the other hand, is like a chess game, only whose complexity is multiplied by an almost-infinitesimally-large number. How do we ever hope to predict it?
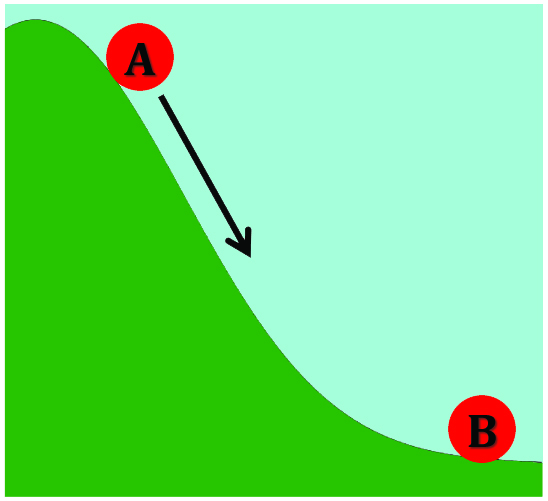
We humans aren’t too bad at holding two objects in our head at one time. Ball A rolls down a hill and hits Ball B which causes Ball B to move. Okay, no biggy. But once we add in a Ball C, then we may start having problems. And heaven forbid we throw into the mix Balls D, E, F, and G!
Very rarely can we identify an issue in biology that’s as simple as Molecule A interacts with Molecule B which causes Molecule B to change its chemical behavior. That’s not to say that adding a given catalyst to a reaction can’t cause changes in chemical behavior. But more often it’s this scenario: Molecule A interacts with Complex B (made up of Molecules B, C, D, E, F, and G) which causes Complex B to change its chemical behavior. But in order to predict how Molecule A would interact with Complex B you already have to know how Molecules B, C, D, E, F, and G interact with each other especially if these subunits vary. Strictly from a probalistic calculation, the potential complexity of Complex B could be represented by:
B x C, B x D, B x E, B x F, B x G, B x C x D, B x C x E, B x C x F, B x C x G, B x D x E, B x D x F, B x D x G, B x E x F, B x E x G, B x C x D x E, B x C x D x F, B x C x D x G, B x D x E x F, B x D x E x G, B x E x F x G, B x C x D x E x F, etc. etc. etc.
In other words, 720 (6 x 5 x 4 x 3 x 2) possible combinations of interactions which we could feasibly study and which, chemically, could each have divergent outcomes. Now add Molecule A and I think you might grasp how even the simplest of biochemical interactions can become overwhelming. You can undoubtedly generalize Complex B as a single unit, but in cases in which the constituents vary you need to know at that given moment in time what subunits comprise the larger unit.
Have your eyes gone crossed yet? 😀
I like to remind people, laymen and scientists alike, how complex biology is. The purpose of this isn’t to overwhelm and stalemate scientific progress but hopefully to remind people to be cautious with their hypotheses and avoid broad generalizations that rest upon a Ball-A-hits-Ball-B foundation. Let me give an example:
Genetics. We love the idea that a single gene codes for a single gene product (usually a protein) and that if a mutation occurs within that gene, that will translate into a change in the gene product. We then also like to take that construct and then stretch it even further to say things like, “Because Single Nucleotide Polymorphism (SNP) B occurs in Gene A more frequently in people who do really well on intelligence tests, that means that this mutation in Gene A is causally linked with intelligence.” This kind of data interpretation happens all the time, and this in fact is taken from a recent example of research performed on the HMGA2 gene and intelligence. Here’s a sample report from BioNews:
“[HMGA2] affects the overall size of the brain and links to intelligence. People with a small change in this gene had larger brains and performed slightly better on IQ (intelligence quotient) tests in studies.”
And here is a quote from a blogger who’s taken it even a step further:
“A mutation in a single gene, [HMGA2], has been identified as being responsible for intelligence. [HMGA2] has been found in people with larger brains and has a positive correlation with the level of IQ. But if a large brain is associated with high IQ and the likes of Einstein apparently had a smaller than average brain surely his ingenuity must be down to the nurtured brain as supposed to this genetics, bearing in mind that the mutation in [HMGA2] only increases IQ by an average of 1.3 points. The high IQ in Einstein’s brain must therefore be due to other factors including the nurturing of this non-mutated [HMGA2] gene to behave as if it were in a larger brain to encourage the shift above the average IQ.”
Obviously if there’s any kind of relationship between a gene mutation and a gross phenotype there’s a causal link right? In the world of Ball-A-hits-Ball-B that would be true, and if only it were. But let me give you a single example of how a given gene mutation can have a relationship with a larger phenotype without being causally associated with it. In fact we’ll use a hypothetical (though feasible) example involving big brains:
The molecule, PTEN, sits in an interesting place within the cell. Not only does it help suppress cell proliferation and growth, it also plays additional roles in DNA stability and repair processes. Now let’s just say that the HMGA2 gene for whatever reasons is inherently less stable than your average gene. And let’s say that the activity of PTEN is mildly suppressed during prenatal brain development (maybe a genetic proclivity or an environmental suppressor, doesn’t matter). This subsequently leads to increased proliferation in your neural progenitor population, leading to more neurons and bigger brains. Because you’ve suppressed PTEN this may also lead to increased occurrences of mutations due either to instability or poor repair. Because HGMA2 is a more likely target for mutation than your average gene, on average it will be mutated more frequently in this scenario. But does that mean that the HGMA2 mutation led to the increase in brain size? No, the primary cause in this instance was the reduced PTEN activity. What this scenario actually resembles is this:
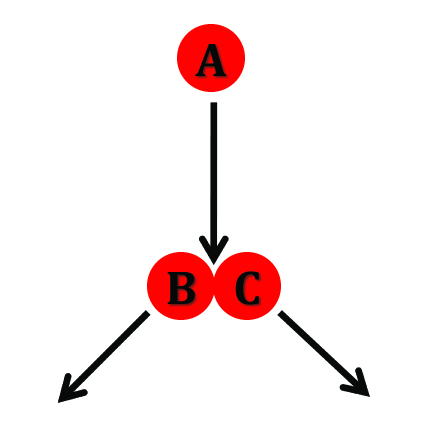
In the above scenario, Ball A is PTEN while Balls B and C are brain growth and the HMGA2 gene. In the case of HMGA2, brain growth, and intelligence the only thing we can be certain of is a relationship, not causation. And in fact, what is currently known about the functions of HMGA2 don’t immediately reveal any obvious relationship to neural proliferation. Here is what GeneCards has to say about it:
“This gene encodes a protein that belongs to the non-histone chromosomal high mobility group (HMG) protein family. HMG proteins function as architectural factors and are essential components of the enhancesome. This protein contains structural DNA-binding domains and may act as a transcriptional regulating factor. Identification of the deletion, amplification, and rearrangement of this gene that are associated with myxoid liposarcoma suggests a role in adipogenesis and mesenchymal differentiation. A gene knock out study of the mouse counterpart demonstrated that this gene is involved in diet-induced obesity. Alternate transcriptional splice variants, encoding different isoforms, have been characterized.”
As it says, HMG proteins are essential components of the enhancesome, which is a complex that binds to enhancer regions of DNA and upregulates or “enhances” the rate of transcription. Obviously these will be involved in brain development, but they’ll also be involved in every other kind of tissue. At first glance, there is nothing overtly obvious linking HMGA2 to neural proliferation.
Now, I want to state categorically that I’m not saying certain types of HMGA2 mutation don’t affect brain size and subsequent changes in intelligence; it could well happen. What I am saying is that we’re getting far too ahead of ourselves drawing causal inferences without even knowing what this gene product does in such a scenario. And in the case of known SNPs in this gene, we don’t know how those mutations ultimately effect– or don’t effect– its gene product(s). We need to answer these questions first before we begin to address whether it leads to changes in brain tissue formation and ultimately behavior.
It’s all too easy to get sucked into Ball-A-hits-Ball-B thinking. One thing which I find helps me from jumping ahead of the data too frequently is to remind myself of specific scenarios like the PTEN one above. It keeps me skeptical, it keeps me thinking and retesting my assumptions. And, in short, it helps keep me honest.
I’d like to invite any readers to proffer their examples of anti-Ball-A-hits-Ball-B thinking within the comments. The more the better!

Where to start? “If ‘A’ —-> (then) ‘B’ ” is so common that the possibilities leave me paralyzed.
But I’m reminded of Daniel Kahneman’s book, Thinking, Fast and Slow (Farrar, Straus & Giroux / Winner of the National Academy of Sciences Best Book Award in 2012)
from the publisher’s page & description:
…” In the highly anticipated Thinking, Fast and Slow, Kahneman takes us on a groundbreaking tour of the mind and explains the two systems that drive the way we think. System 1 is fast, intuitive, and emotional; System 2 is slower, more deliberative, and more logical. ” … ( http://us.macmillan.com/thinkingfastandslow/DanielKahneman )
so there is something of “System 1” thinking in the impulsive urge to pounce at first glance upon the idea of cause–> efffect.
It’s everywhere around us (because it’s in all of us.) Kahneman explains that we are all subject to both “System 1” and “System 2” thinking habits. Our best defense is an informed awareness of the modes and occasions which are common to them. In the right circumstances both are useful and both are troublesome in the when misapplied or allowed to run on “auto-pilot-think.”
Sorry for the delay in response; work has been rather insane the last couple days. I agree with your thoughts. A > B thinking certainly has it’s place, but, like you suggest, it’s all too frequently OVER-applied. What shocks me most is not that many laypeople think in this way but that scientists– those who are supposed to be, on average, more skeptical and detail-oriented– still over-use A > B thinking. (And I’m not excluding myself from that necessarily. I’m sure I use A > B way more often than necessary.)
It’s extremely frequently misapplied, yes, indeed.
In this case, however, the you and the other scientists are in the same boat with the rest of we, the average lay-people.
There’s simply no training–or, as they say, “no known ‘cure’ for this” , ;^) — no “fix” that can definitively eliminate or even significantly reduce this natural tendency in us. That is, indeed, one of the points that most impressed D.K. in the course of his research. He found that judges, lawyers, scientiests, engineers, –all sorts of highly-trained professionals–are just as subject to these habits of thought as anyone else. The implications are quite serious. Think of the possibilities: CEO’s, or criminal court judges–indeed, think of the Supreme Court’s Justices–they aren’t immune at all!– or Wall Street market-traders who are handling multi-million or multi-billion dollar trades in stocks, bonds, treasury bills and notes and all sorts of derivatives.
For those who are facing a judge’s criminal court sentence, consider: research data show that decisions taken (whether by a single judge or a tribunal “en banc”) after lunch rather than just prior to lunch are significantly more temperate in character–IOW, better your sentence is handed down soon after lunch than before. In “dispensing justice,” there should be no such place for that kind of discrepancy. But it’s there (along with numerous other types just as terrible)–in any system which doesn’t have means and measures to take it into account.
Please note: Another and related text is Robert Trivers’ The Folly of Fools: The Logic of Deceit and Self-Deception in Human Life. It came out at around the same time as Kahneman’s book and its no less brilliant. Each author’s work and book complements the other’s.
er, that should’a been ,
“in the same boat with the rest of us average lay-people, “…
or, “in the same boat with we average lay-people …
sheesh! Mah grammar!
Yeah, I’d have to say that at least the earlier stages of scientific training don’t really teach so much how to hypothesize effectively but moreso how to carry out experiments. It can have a roundabout way of improving some analytical skills but I think much of that is left to the ingenuity of the budding scientist, not so much due to the structure of a program. Thanks for the additional refs. And feel free to break any grammatical rules you wish. I’m no grammar nazi. 😉
Some recent thoughts and reading regarding
Technology’s part in our thinking traps and errors—
Yesterday, I attended a day long series of round-table discussions aimed at the interests of the general public with, for main title, “The Year in Review, as seen by …the Sciences”. There’d been a similar conference held there and entitled, “The Year in Review …seen by Philosophy” (or Philosophers) and another, still to come, from the point of view of “Historians.” (Though, in fact, there was no round table, just the participants seated in an array on the stage of the grand auditorium of the Sorbonne—a beautiful semi-circular auditorium with a fresco by Pierre Puvis de Chavannes, “le Bois Sacré” (1887-1889) (“the sacred wood”) as the background above the stage. (Links to photos of the hall and the artwork: http://www.larousse.fr/encyclopedie/image/Laroussefr_-_Article/1313808 ;
http://www.sorbonne.fr/location-grandamphi.html ;
http://paris-bise-art.blogspot.fr/2011/11/le-grand-amphitheatre-de-la-sorbonne.html )
Yesterday’s program offered a series of six discussions of 75 minutes on
“The Case Pistorius: the human body augmented or repaired?”
“Curiosity on Mars: Is Space the future of Mankind?”
“Melting Glaciers: Does indifference protect us from Global Warming?”
“Nuclear Power, an industry of the future? What chain-reactions?”
“Higgs Boson, Genetically-Modified Organisms, Shale-Oil Gas…Is Science the problem or the solution?”
“Nobel Interview: Serge Haroche, Nobel Laureat for Physics*, 2012”
Running just under the surface of each discussion, as though there was some sort of earth-tremor rumbling beneath everyone’s feet, were hardly disguised concerns, on the parts of both audience and speakers, about where science, especially as its work and discoveries relate to the technology, is taking us as a society and how as both individuals and as societies, these trends are shaping us, our ways of thinking and our habits of living.
In the last segment, Serge Haroche spoke in detail about how in his field research has, in the course of the past twenty years, come more and more under the authority of agencies which impose reviews and call for justifications for science research—often by groups which are outside the community of researchers–and expecting researchers to justify their work by its prospects for some eventual pay-off in knowledge and technology. He presented an argument for a reform of such an approach to evaluating present or prospective research work, explaining that it springs from a fundamental lack of confidence in professional science research to competently direct its work. Along the way, he had a word implicating the transformations of the world of business and finance since the administrations of Ronald Reagan and Margaret Thatcher, with their supposedly “bottom-line” view of the worth of all human endeavors, science research not excepted.
But he had little or nothing to say about the, to me, crucial connection which all sorts of technology has had in the rise of the kind of expediency-driven view of human affairs nor how such a view is contained, implicitly and inherently, in the technologies themselves.
Researchers have found themselves caught in the web of technology’s inherent value-systems—in which there is a fundamental disregard for anything human in character. Technology doesn’t and simply cannot take any account of human-based needs, interests, considerations, or priorities because its foremost driving imperatives are contrary to these—they are based entirely in considerations which turn on issues of expediency and efficiency in the narrowest sense. Anything which has a lower or lesser value in efficiency is, by definition less worthy of a place in the order of things. Human needs and interests are, by this view, to be judged and measured according to their likely-product’s fitness, their appropriateness to such efficient and material, quantified ends.
So, research in some area which hasn’t an obvious justification by practical measures—read, “financially justifiable”—is automatically suspect and, therefore, at risk.
All of that resonated strongly for me because it tracks closely some of my current and recent reading, including a very recent text (in French, “Global Burn-Out” ( the original French publication adopts the English terms for its title) by Pascal Chabot, (P.U.F., Paris, 2013) and, related, L’engrenage de la technique: essai sur une menace planétaire by André Lebeau, (Editions Gallimard, Paris, 2005).
In his book, Pascal Chabot presents as a case example the experiences of and the resulting book by Matthew B. Crawford, (originally, Shopcraft as Soulcraft: An Inquiry Into the Value of Work, (2009, Penguin Press ; Éloge du carburateur, (in its French edition, La Découverte, Paris, 2010) who poignantly describes his journey from recent graduate to the world of market-interest-and-technology-driven employment. (For a brief exposition of the highlights, see Crawford’s New York Times Magazine essay, “The Case for Working With Your Hands,” May 24, 2009)
(from (in Part 2: La Machine Burn-Out, page 51, “ To Have Done With Perfection”) at page 57, Chabot writes, “ Burn-out is the mirror of an adaptation become absurd and frustrating because it has for its end only itself. It’s the trap of an unattainable perfectionism. The story of Crawford and his (work) can be read as the attempts by a person of good-will to acclimate himself to the best degree possible to constraints which, each time they become bearable, evolve again toward greater difficulty.” (my translation; my note: That is, every time Crawford met his work-quotas, he found those quotas adjusted, increased, so that he could never reach a sustainable level of satisfactory output.)
One of the other things noticed and actually commented on in the course of the discussions was how democratic processes have become rendered futile and inoperative, unresponsive to the plainly obvious needs and interests of ordinary average citizens, how, instead, the institutions of democracy are now the corrupted prisoner of the same technology-driven priorities which favor income-based and quantifiable accountancy measures, and which serve the same centralized corporate needs and interests which are diametrically opposed to the human-scale and humane needs and interests of the public which is now shut out of any effective part in the society’s ordering of priorities—however profoundly these are imposed on that public.
____________________________________
* Nobel Prize for Physics, 2012, shared with David J. Wineland; http://www.nobelprize.org/nobel_prizes/physics/laureates/2012/ )
I would truly love to visit the Sorbonne, it looks incredible, and its history fascinating.
For all you say about research needing financial justification, it’s so very true. Researchers are used to writing grant after grant proposal, attempting to justify the usefulness of the data which may potentially be derived from any given study. If one is interested simply in learning more about a topic in biology and it is not something which lends easily or immediately to some sort of medicinal intervention, it can be very difficult to get funded. Even basic research must often present the “usefulness” of any given data set. On the one hand, biological research defines medical intervention and human health is deemed more important and therefore more fundable. Heck, philosophy has often been chided as only “mental masturbation” and so I suppose some of the sciences which are also “knowledge for knowledge sake” could fall under the same criticisms. On some level, I understand the demand for pragmatic foci. On the other hand, it is very difficult to predict what will be useful and what won’t and so to regulate intellectual freedoms in such a way may prevent important discoveries as well.
I don’t quite know what it is, but I feel like scientists themselves have changed. Where today do we have the modern equivalents of the great British or French naturalists? Yes, competition has increased over the centuries because it’s not only the educated rich who have the means to study life. Now anyone can, provided they can get the degree. Which is wonderful that so many minds have access to our knowledge base and can squeeze every bit of information from it available. But the quality of the scientist has changed; I don’t see the passion, the desire to understand, the craving to dig and dig until you hit rock and can’t dig anymore. Perhaps it’s that the majority of people never know this kind of passion and since so many are scientists, the mediocrity and– dare I say– boredom is inevitable. To some, to many perhaps, it is just a career. But perhaps also this pragmatic approach to science and discovery is an antidote to passion. Writing grant after grant, occasionally getting funded, most times getting rejected; writing publication after publication with so many reviewers demanding infinite revisions; even the hassle of finding the “right” journal that shares a similar POV as yourself– and heaven forbid there isn’t one! The the passion inherent to mental masturbation, knowledge for knowledge sake, can push the boundaries of what we know and lead to awesome discoveries. Science used to be so romantic… Where has the romanticism gone?
I don’t have all the answers to such an interesting set of philosophical questions about your profession. But these questions are very much among the matters that so occupy my own thoughts and interests in reading and in trying to understand better what is going on in the world–inside and outside of the sicence professions. Bertrand Russell, for example, wrote an interesting essay concerning the faith with which science’s early prioneers began and which its practitioners since then have needed and drawn upon but which, even at the time he wrote— over 80 years ago in this instance, if I recall correctly– was already, by his account, showing indications in the advanced Western industrial world of suffering from a flagging in this basic faith among some scientists. (see his anthology, The Basic Writings of Bertrand Russell, in (I think) the chapter headed : The Philosopher of Science ; again if my memory serves me here)
But I think that technology’s developments, esp. since Russell’s essay, and the kind of internal logic of technology to which I allude above, have had as much or perhaps more powerful catalysing effects on this sapping of faith in and enthusiasm for the promise of science as a bringer of light and understanding to the world. It’s a great shame. And yet, everything good and useful about science as an intellectual endeavor is just as valid today–and just as needed–as it ever was. But because of a variety of factors which are mainly sociological and moral in origin, science has taken some hits, has suffered, for the weakenesses and failings that are inherent in people–all people, scientists and non-scientists alike.
We need better understanding in so many areas and better moral insights and better, more open and honest dealings among classes and nations of people. Science has so much to offer in aid of all of these things. It’s a shame that in pushing ever further the limits of knowledge, something in faith and enthusiasm for science has been lost among some–, perhaps has been in a sense “paid”, in a kind of action-reaction trade-off.
Come and visit the Sorbonne when you can; and while you’re at it, don’t overlook the Institut Curie, or the Curie’s laoboratory, near the Panthéon, where the famed couple, Pierre and Marie Curie did their laboratory research.
>> It’s a shame that in pushing ever further the limits of knowledge, something in faith and enthusiasm for science has been lost among some–, perhaps has been in a sense “paid”, in a kind of action-reaction trade-off. <<
Well put. Times have definitely changed. I just thank the fates I'm an absolute romantic at heart, in the classic sense. Maybe more than just my writing has been shaped by the fact that my initial studies were in English and creative writing and only eventually moved into research. I studied poetry initially. Eliot, Brecht, Rilke, Cummings, Poe… I love them all. Perhaps the arts could breathe fresh life into science?
re: ” Maybe more than just my writing has been shaped by the fact that my initial studies were in English and creative writing and only eventually moved into research. I studied poetry initially. Eliot, Brecht, Rilke, Cummings, Poe… I love them all.” …
I agree completely with that view. Your earlier literary studies are a great advantage and a great value–one not “seen” or understood in our foolish times. You’ll always have the benefits of those studies in everything that you do. I think that this aspect of your knowledge and its manner of shaping your intellect is a very great part of how and why your science blog here is so wonderfully different and better than what’s typically found in blogs about science
“Perhaps the arts could breathe fresh life into science?”
They could–provided, of course, that more than a few outliers of science took an interest in them. It’s funny, but, thinking of scientists who are regarded as a giants, a stand-out geniuses among their peers, frequently–in the past, at any rate, they were also interested in the arts–literature, history, poetry among others. Those interests informed and improved his or her intellect.
To correct my faulty memory: Russell’s essay, mentioned above, is not after all found in the anthology, The Basic Writings of B.R.. Instead, it’s his essay, “Is Science Superstitious?”, from his collection, Sceptical Essays, first published in 1928.
…”The scientific creator, like any other, is apt to be inspired by passions to which he gives an intellectualist expression amounting to an undemonstrated faith, without which he would probably achieve little. The appretiator does not need this kind of faith; …. As civilisation becomes more diffused and more traditional, there is a tendency for the habits of mind of the appreciator to conquer those who might be creators, with the result that the civilisation in question becomes Byzantine and retrospective. Something of this sort seems to be beginning to happen in science. The simple faith which upheld the pioneers is decaying at the centre.” So wrote Russell in 1928.
Your background in literature affords you a kind of innoculation against this loss of passion and faith. Others, without it, are much more given to the ennui of having little of substance at their own spiritual centres–and science alone doesn’t satisfy this inner void consistently for them.
Our own times, strangely, are indeed extremely Byzantine in character but, instead of their being retrospective–as Russell describes the malaise– our times are at once naïve, insecure, nostalgic and yet desperately focused on a confused and unenlightened present and fearful of and anxious about the future. Part of this anxiety is due to the growing recognition that it is a blind process of technological development–over which no single individual, not even the most exalted scientist or political actor, has any effective influence–is the driving and controlling factor in our lives and our societies.
In all of this, your knowledge of and appreciation for literature and your practice of it, as a thoughtful writer, afford you points of spiritual reference in what, for others, is a terrifying storm of constant and bewildering change.
Plus having that romanticism about science just makes my work so much fun. 😀
Browsing, so arrived here. I call them the Determinista – singularist, cause and effect and mechanistic in approach, as if biology had been put together by Volkswagen in a factory in Wolfsburg.
With four billion years of evolution behind us, there’s room for a great deal of subtle sensitivities and polycentric interactions as you suggest. One of my favorites, as exemplar, is the possible genetic codings associated with the condition(s) described as psychopathy where control sequences or multiple copies of control sequences could well be involved rather than the gene itself.
Another area is “non functional” differences in gene codings at any one allele through a population. ICGO….but not now!
Most definitely. And if you like the idea of large copy number variants’ involvement in psychopathy, you’d probably have some interest in the Jumpin’ Genes section I’ve written, focusing on transposable elements. Many large CNVs shared tight links with these little mobile guys.