I’ve talked a lot about DNA mutations, transposons and retrotransposons, microsatellite repeat sequences in cancer and autism, and even somatic mosaicism on this blog. But I haven’t really talked about all these things in relation to my favorite organ system: the brain.
Now why, above any other system, would I single out the brain? Aside from my own personal bias (–ya gotta admit, the brain is pretty friggin’ awesome), it often displays a surprising level of somatic mosaicism as compared to the rest of the body. As a reminder to readers, and for those new to the site, somatic mosaicism refers to the presence of two or more cells in a single organism which display variations in genotype. Severe mosaicism sometimes in the form of extreme chromosomal malformations can be a sign of cancer; considerable mosaicism in a single gene across tissue types can be indicative of various genetic diseases such as some of the polyglutamine (polyQ) diseases I’ll refer to later; and meanwhile moderate mosaicism is normal and to be expected [1, 2].
LINE-1 (L1) transposable elements are a family of retrotransposons, some of which are still transpositionally active in the human genome. By “transpositionally active” I mean that they are still capable of producing RNA copies of themselves which can be reverse transcribed into DNA and inserted elsewhere in the genome (see image below). Some studies have shown that L1 retrotransposition can account for a certain level of mosaicism in the brain, particularly within the hippocampus, an interesting finding considering the hippocampus produces neurons throughout adulthood [3]. That same study by Baillie et al. (2011) reported a somatic retrotransposition event in the intron of Hdac1, a histone deacetylase which is a known regulator of L1 expression within the human genome, suggesting how a single transposition event might alter the capacity of a given cell to regulate further insertions [4].
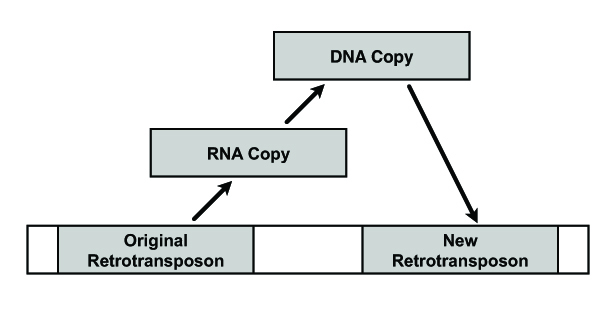
As a segue, retrotransposition usually facilitates the creation of short repeat sequences (microsatellites) within the genome at the points of insertion (e.g., CACACACA). They arise through the act of reverse transcription and are referred to as target-site duplications (TSD). Microsatellites litter the genome, exonically and intronically, and as I discussed last week are prone to inducing various types of mutations within a given gene particularly when the local DNA conformation is under greater tensional strain. There can be global strain upon a chromosome or, more commonly, local strain placed upon it through various binding partners such as transcription factors.

These microsatellite repeats, particularly trinucleotide repeats, have been indicated in a number of diseases, especially relating to the central nervous system. The first trinucleotide repeat disorder to be identified was Fragile X syndrome, a condition which frequently exhibits co-occurring autism and is usually due to an expansion of trinucleotide CGG repeats which ultimately promotes hypermethylation and reduced gene expression [5].
A group of disorders known as “polyQ disorders” are due to CAG repeat expansion within coding regions of certain genes. They are known as “polyQ” because “Q” is the symbol for the amino acid, glutamine, which is coded by the sequence “CAG”. With the expansion in CAG repeats, so too does the protein product exhibit an expanded glutamine tract. Disorders falling under this umbrella category include: spinal and bulbar muscular atrophy (SBMA), Huntington’s disease (HD), dentatorubral pallidoluysian atrophy (DRPLA), and spinocerebellar ataxias (SCA) type 1, 2, 3, 7, and 17. Given the common etiology underlying these varied disorders, for some time it had been a mystery to scientists how glutamine expansion tracts could prove toxic. At least with the SCA type 1, there is evidence to suggest that in some instances the protein product exhibits loss of function and, most surprisingly, at other times as gain of function is observed. But as far as toxicity, it has also been shown that polyQ tracts have a high propensity to aggregate and form cytotoxic neuronal intranuclear inclusions (NII) [6]. Investigations into chaperone complexes which can unfold or disaggregate these polyQ inclusions are promising as a treatment for symptoms of these disorders.
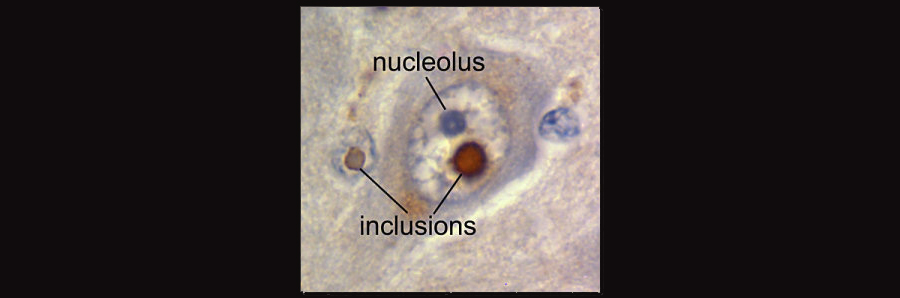
The above image shows a nuclear inclusion from a Fragile X patient with ataxia. Such inclusions are associated with toxicity in some of the trinucleotide repeat disorders. Image borrowed from HERE.
All of these disorders have reported somatic mosaicism within the mutated genes, such that different extremes of expansion are seen in different organ systems. As you might now be wondering given the topic of this blog post, what are the rates of mosaic expansion in the brain? Well, for the majority of the CAG repeat disorders, the brain exhibits comparatively greater expansion than other tissues, with the notable exception of SBMA, which instead shows greater variability in cardiac and skeletal muscle, skin, and prostate [7]. Aside from the obvious, that these conditions tend to be overrepresented by neurologic issues, why might the brain exhibit greater repeat expansions than other organ systems?
Damned if I know.
BUT if I had to take a guess, just a wild guess, outta the blue, put the ol’ thinking cap on and give it a good long Rodin-style try… I would say that these mutated genes 1) lie in regions which have a greater baseline superhelical strain which, upon binding with just the right (or in this case, wrong) partners, promotes alternate DNA conformations which are prone to replication errors and breakage, and 2) the expression of these mutated genes may be key in central nervous system development and/or homeostasis, so that they are constantly being transcribed and therefore made continually vulnerable to this type of mutation.
The brain after all is a very busy system. It consumes vast amounts of resources and never sleeps. Just imagine how much transcription is continually going on and how each transcription event is a risk for mutation.
It’s probably not as simple as this. (When is it ever?) But I have a strong suspicion that transcriptional activity may play a key role in this type of mosaicism. Using Occam’s Razor, it’s the simplest way to specifically induce increased superhelical tension on a target gene and induce mutation. And as scientists plod on in their studies, one day we may find out whether this prediction is true. I know I’m keeping my fingers crossed. 😉


Another fascinating lesson. Thank you!
“Aside from the obvious, that these conditions tend to be overrepresented by neurologic issues, why might the brain exhibit greater repeat expansions than other organ systems?”
I’m curious– are the greater number of repeat expansions found in the brain taken as a proportion of the organ’s (let’s say average) total cellular size? That is, thus, per millions or billions of cells, the brain presents higher instances of these repeat expansions?
————————————————-
By the way, in Scientific American (May, 2009, Cover article: “What Makes Us Human?”), the researcher, Katherine S. Pollard, writes about, among other things, the “HAR1” “Human Accelerated Region 1” of the brain, with its higher frequency of mutatiuons. I bet you’d find her work interesting if you don’t already know about it.
(from Wikipedia, on HAR1: “In molecular biology, Human accelerated region 1 (highly accelerated region 1, HAR1) is a segment of the human genome found on the long arm of chromosome 20. It is a Human accelerated region. It is located within a pair of overlapping long non-coding RNA genes, HAR1A (HAR1F) and HAR1B (HAR1R).[1]”
link: http://en.wikipedia.org/wiki/Human_accelerated_region_1
http://www.scientificamerican.com/article.cfm?id=what-makes-us-human
http://gladstoneinstitutes.org/scientist/pollard
That’s really hard to say. I suppose the general size of the organ, indicating the level of its developmental proliferation, could affect numbers of specific expansions. But moreso, the brain appears to experience continued repeat expansions even in mature neurons (as do other cells from other organ systems, but the brain seems to be somewhat unique in terms of certain microsatellite expansions within particular genes). And each neuron is potentially unique; you kind of have to imagine each cell as having a common genome but that there are minute yet distinct variations by cell. When someone is genotyped, say by taking a blood sample, most of these small percentage variants tend to be “washed out” in the process (the process of amplification naturally favors sequences that are in the majority while the remainder just end up being background noise). This can make it look like all the cells in the person’s blood have the same genome, when in fact each cell usually has small variations. In the case of cancer, these can be even larger variations. Likewise, in the case of Huntington’s Disease, the extent of CAG repeat expansions in the Huntingtin (HTT) gene are 1) tissue specific (largest expansions observed in the brain), 2) size specific, in that the size of the expansion the person is actually born with ultimately affects its stability so that longer expansions are less stable, and 3) age specific, i.e., greater expansions occur over time. It is believed that this last factor is the reason Huntington’s occurs so late. The mutations are there but they become progressively worse such that, eventually, numerous expansions exist and the protein contains a high level of glutamine, promoting cellular toxicity.
Interesting! Thank you! Your comments prompt other thoughts and questions. So, until your patience runs out, here are a few more–
Is there a current favorite theory as to what basically drives greater/lesser superhelical tension in the first place–as a general phenomenon–quite apart from where its found in chromatin?
What does laboratory experience show when it comes to superhelical tension and related mutation, if any more or less occurs in chromatin when observed outside the body’s living environment? Similar rates and kinds of effects?
And RE: ” …lie in regions which have a greater baseline superhelical strain which, upon binding with just the right (or in this case, wrong) partners, promotes alternate DNA conformations which are prone to replication errors and breakage… ”
this, perhaps wildly naive, question:
in a laboratory tissue sample (of any convenient type) which has high “PolyQ” disorders, is there any reason to suppose it might be interesting to attempt to artificially introduce other trinucleotide repeats as a means –excuse me if the concept is simply completely ridiculous–of “restoring a (numerical/or chemical?) ‘balance’ “, in a manner of speaking?
Hey, proximity, sorry for the delay in reply. Just finishing Mother’s Day festivities here. As for your questions, I’ll give it a shot:
1) You ask about a theory which attempts to explain superhelical density. Well, since biochemical molecules are not just about charges but about the overall shape of a given molecule itself, the changes in the twist of the local DNA double helix are resultant not only of the native tendencies of the DNA itself but also the changes in local shape that its binding partners induce. These would include any number of regulatory proteins, RNA, metal ions, carbs, and lipids which find a “platform” in a specific tertiary (3D) DNA sequence. Just consider some of these binding partners to act like catalysts in a reaction, in this instance prompting a change in the degree of twist in the DNA local DNA molecule. I don’t know precisely if that’s what you’re asking but hopefully my description might help in any regards.
2) The second question, it sounds like you’re asking whether petri dish (in vitro) research finds similar changes in torsional strain as is found in vivo (life). I’m not absolutely certain as far as level of superhelical density in vitro vs. in vivo; that’d really take going through the research paper by paper and tallying. Essentially, DNA will behave differently with different binding partners. I suspect that if one is investigating via cell culture in which the DNA is in a relatively similar nuclear environment as in the body, the differences are probably fewer. However, since a cell alone is not the same as a cell within a community, some of the active pathways may be different and therefore nuclear binding partners may differ as well. Larger trends would probably still be applicable across both situations.
3) As for you last question, it sounds more like a reference to gene therapy. Although in the case of many trinucleotide repeat disorders, you wouldn’t want to increase the number of repeats are add other repeats into the same gene region. But promoting trinucleotide deletions may be a thought– albeit a difficult one to promote, and would probably end up being sequence specific.