This week I’ll continue reviewing more forms of high-association syndromic autism. Some of the conditions I’m writing about today include not only targeted gene mutations, as in the case of Tuberous Sclerosis, but also deletion and duplication syndromes. These deletions and duplications include larger bands of chromosome and span multiple genes, making their etiology more complex than single gene conditions.
Isodicentric Chromosome 15 Duplication Syndrome
Bands q11.2-q13.1 on the long arm of chromosome 15 are a considerable hotspot for autism. Duplications along this stretch of DNA are the cause of Isodicentric Chromosome 15 Duplication Syndrome, shortened idic(15), and meanwhile deletions give rise to both Angelman (AS) and Prader-Willi Syndromes (PWS) which will be discussed in coming sections. A staggering 92% percent of cases of idic(15) also fulfill criteria for autism. While symptoms vary, most individuals present with mild-to-profound intellectual disability. Poor muscle tone is also common and tends to lead to delays or impairment in crawling and walking, as well as problems feeding. The majority of cases usually present with seizures, often within the first year of life, and strabismus occurs in a large minority of cases.
The 15q11-13 stretch of DNA is considered generally unstable due to the presence of duplicons or segments of duplicated DNA [1]. In short, these duplicons can promote misalignment during DNA replication that can create a number of breakpoints or vulnerable spots within the region. These breaks can lead to either further duplications as in the case of idic(15) or deletions as in AS and PWS. Scientists have determined that this locus houses four breakpoints, and the duplications which promote instability tend to include duplicated segments of DNA from the HERC2 and GOLGA8E genes which disrupt the region. These duplicated segments of HERC2 and GOLGA8E are referred to as “pseudogenes”.
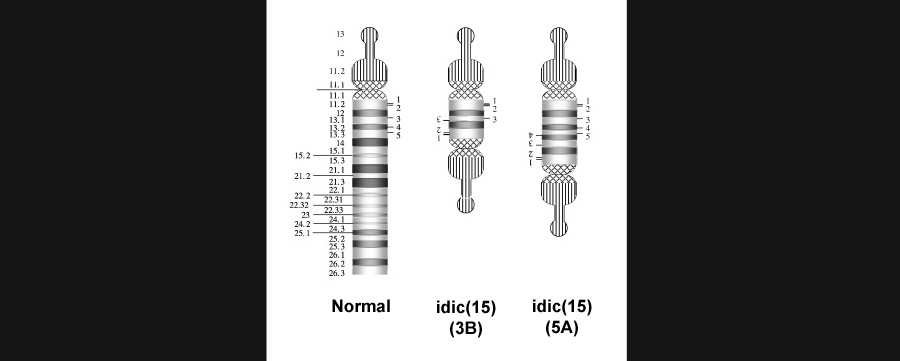
Image of a normal 15q11-13 region, followed by two different forms of idic(15) duplication and inversion. Borrowed from here.
The 15q11-13 region exhibits parent-of-origin effects, which means that the same mutation can have different results depending on whether you inherit it from your mother or your father. This usually has to do with different trends in epigenetic regulation, e.g., methylation patterns, between maternally- and paternally-derived genes which leads to variations in specific gene expression. In the case of idic(15), the duplication/inversion usually is inherited from the mother. And just as with Down Syndrome, risk for idic(15) increases with maternal age, although comparatively idic(15) is a much rarer disease estimated to occur in about 1 in 30,000 births [2].
Scientists have found that when when idic(15) duplications occur around the 15q11-13 region but don’t actually include the Prader-Willi Syndrome/Angelman Syndrome (PWS/AS) region, the affected person often shows no traits indicative of the gross mutation and may be phenotypically normal. And while it is believed that many of the traits of AS are due to UBE3A gene deletion or mutation on the maternal chromosome and PWS to the SNORD116 noncoding region (see PWS below), the cause of idic(15) has yet to be identified.
Nevertheless, UBE3A may be involved in its etiology, as well as SNORD116, and some research has also suggested that effects on gene expression of GABA receptor-related genes, of which there are several in this region, are altered in idic(15) [3]. Since GABA is the primary inhibitory neurotransmitter within the brain, decreased GABA receptor production reported in idic(15) may help explain their propensity towards seizures.
In contrast to AS, it is believed that while gross duplications are the ultimate causes of these syndromes, these mutations in fact drive changes in methylation patterns and thus gene expression within the 15q11-13 region. Therefore, epigenetic changes of methylation seem to be the direct promoters of idic(15).

A lovely young lady named Corrina who has idic(15). Read here for more on her story.
Angelman Syndrome
As with idic(15), Angelman Syndrome is a 15q11-13 disorder. Approximately 63% of those diagnosed with AS have comorbid autism. Symptoms include developmental delays, intellectual disability, coordination and balance problems, seizures, language impairment, and microcephaly. One thing which characterizes many of those with AS are their happy smiling demeanors.
As mentioned earlier, AS is caused by loss-of-function of the Ubiquitin-protein ligase E3A (UBE3A) gene. Most cases are due to large regional deletions which include the gene, but a smaller percentage occurs because of a mutation within the gene which impairs its expression or functionality.
UBE3A acts as a ligase for ubiquitin, which means that it helps attach ubiquitin to various molecules. Ubiquitin performs as a multifunctional tag, with its best-known role as a marker for protein degradation or destruction. So whenever a protein is marked with the appropriate amount of ubiquitin, the cell knows to destroy that protein. This is part of the disposal-waste system of the cell in which it can get rid of old or misfolded proteins. If such proteins are not “ubiquitinated,” they can build up and cause problems for the cell. In people with AS, since most of the cells of their body can rely on the father’s functioning copy of UBE3A, primarily the brain which relies on the mother’s missing or mutated copy shows signs of the condition.

Colin Farrell with his son, who has Angelman’s and a form of cerebral palsy.
Prader-Willi Syndrome
This syndrome, as with idic(15) and Angelman’s, is associated with the 11q-13 region of chromosome 15. And like AS it is usually caused by a deletion of genetic material from the region. Unlike AS, however, this condition is usually paternally inherited for a similar reason that AS is maternally inherited: the genes which cause Prader-Willi (PWS) inherited by the father are usually the one’s expressed, while the mother’s are epigenetically silenced. If the genes inherited from the father are missing or malfunctional, PWS occurs.
A significant minority (23%) of people with PWS also fulfill criteria for autism. The primary symptoms of PWS include: mild-to-moderate intellectual disability, learning disabilities, developmental delays, growth delays, poor muscle tone, hypogonadism, and delayed or incomplete puberty. Facial features can include almond-shaped eyes, a narrow distance between the temples and a narrow nasal bridge, and a thin upper lip and down-turned mouth. While infants with PWS tend to have problems early on feeding and gaining weight, by childhood those with PWS suffer considerable challenges with overeating. In fact, this symptom of the condition tends to be one of the most challenging due to the health problems which often accompany morbid obesity.
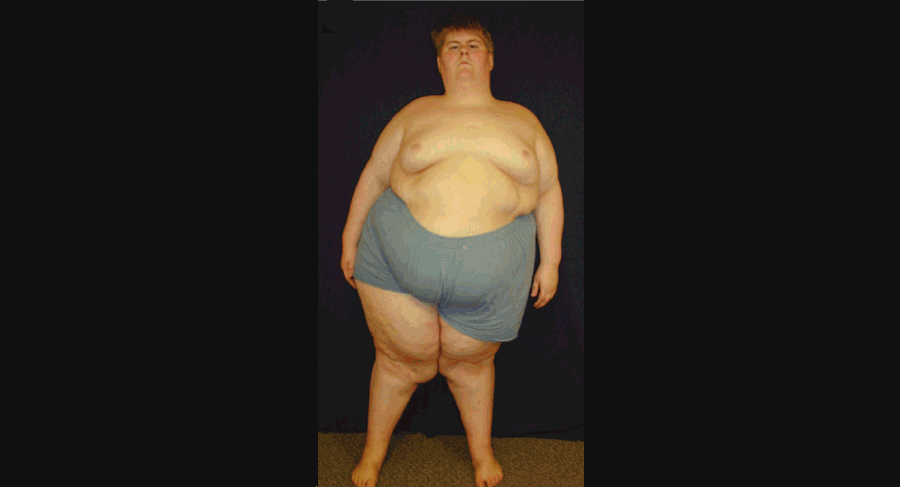
A young man with Prader-Willi Syndrome. Image borrowed from here.
PWS is caused by deletion of SNORD116, a noncoding RNA normally highly expressed in the brain that helps to modify small nuclear RNAs (snRNA). Though scientists know that the loss-of-function of SNORD116 is the cause of PWS, it is still uncertain precisely how its loss leads to the condition because, though its function is clear, it has many different kinds of snRNAs which it modifies, making for a very complex picture.
Tuberous Sclerosis
Autism occurs in 16-60% of those with Tuberous Sclerosis (TSC). TSC is primarily characterized by benign (noncancerous) growths throughout the body, including the brain. Intellectual disability, learning disabilities, behavior problems, and seizures are all common. Tumors on the skin are extremely common, as well as under the fingernails. Benign tumors developing especially within either the brain or the kidneys can prove life-threatening.
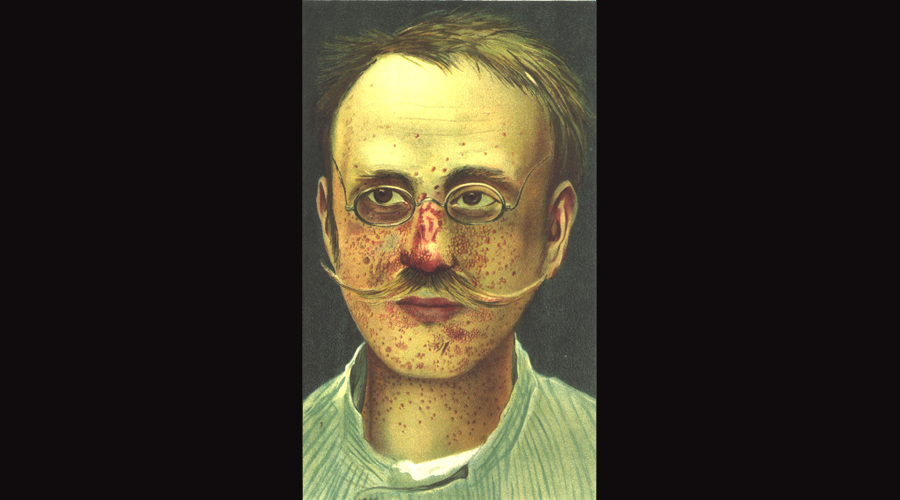
A picture of a patient with TSC from Munich, 1903.
TSC is caused by loss-of-function mutation in either the Tuberous Sclerosis Complex 1 (TSC1) or Tuberous Sclerosis Complex 2 (TSC2) genes which lie at chromosomes 9q34 and 16p13.3 respectively. TSC1 codes for the protein, hamartin, while TSC2 codes for tuberin. Both of these proteins are tumor suppressors and negatively regulate the mTOR pathway, a growth-related pathway within all cells. People with TSC usually have one copy of either TSC1 or TSC2 that isn’t working. While this alone won’t cause tumor growth, it increases the person’s risk. Just like with cancer, once one tumor suppressor goes, it’s all the more likely that another one will follow suit because minor increases in growth increase the likelihood of mutation. In this case, tumors eventually form in TSC in cells in which an additional mutation occurs in the other remaining TSC gene.
Fragile X Syndrome
Fragile X Syndrome (FXS) is the most common “rare genetic disorder,” occurring in approximately 1:4,000 male births and 1:8,000 female births. Approximately 20-60% of those with FXS meet the criteria for autism as well. Interestingly, carriers of the FXS premutation are also more likely to develop autism, ranging from 10-15% in males and 5% in females. Symptoms can range considerably depending on the severity of the condition, with some individuals falling within normal (perhaps a few even in gifted) ranges, meanwhile others may be severely intellectually disabled. Many exhibit developmental and language delays and, symptomologically, males are usually more severely affected than females. Males are also more prone towards characteristic facial anomalies such as long faces, large ears, and a large forehead.
A little boy named Adam who has Fragile X Syndrome. Image borrowed from here.
FXS is a repeat expansion syndrome. The Fragile X Mental Retardation 1 (FMR1) gene on chromosome Xq27.3 houses a CGG repeat DNA sequence. In most people, this triplet is consecutively repeated 5-40 times (e.g., CGGCGGCGGCGGCGGCGG). In carriers of the condition, called the “premutation,” this CGG sequence is repeated approximately 55-200 times. If someone inherits 200+ repetitions they develop the full syndrome. This mutation doesn’t mutate the gene itself but instead effectively turns the gene off through increased methylation. Heavy cytosine (C) content binds methyl groups which lock down the gene and prevent its expression, so while the coding region of FMR1 is normal, it can’t be used.
FMR1’s protein product, Fragile X mental retardation protein (FMRP), is known to be a modulator (repressor) of protein translation. It also binds RNA and seems to play a regulatory role in the trafficking of RNAs out of the nucleus and into the cytoplasm of the cell. As a regulator of both RNA trafficking and protein translation, its effects are probably numerous on cell metabolism, growth, mitosis, differentiation, and motility.
What Do These Conditions Have in Common?
Let’s review what we know and don’t know.
- Idic(15) is the result of a duplication/inversion of the Angelman/Prader-Willi locus on the long arm of chromosome 15. While we don’t yet have a known gene or group of genes that are the cause of idic(15), we do know that the causal gene or genes lie within the PWS/AS-region. A list of possibles include UBE3A, the snoRNA-related noncoding regions like SNORD116, and several of the GABA receptors.
- AS is the result of loss-of-function of the UBE3A gene, which is involved in the degradation of proteins within the cell.
- PWS is caused by the loss of SNORD116, a noncoding RNA highly expressed in brain that helps modify snRNAs allowing them to function properly.
- TSC is caused by a loss-of-function mutation in either the TSC1 or TSC2 genes, which code for the proteins hamartin and tuberin respectively. Both of these proteins regulate activity of the growth-related pathway, mTOR. A loss of one copy of either of these genes leads to increased risk for tumor formation.
- CGG repeat expansion within the FMR1 gene causes hypermethylation of the gene and prevents its expression, causing FXS. FMRP is a negative regulator of protein translation and trafficking of RNA out of the nucleus.
Wow, this one’s a toughy. Problems with protein degradation in AS which could feasibly lead to issues of cellular toxicity may harken back to last week’s review of Smith-Lemli-Opitz Syndrome‘s problems with hypercholesterolemia and toxicity. Meanwhile, PWS’ dysregulation of snRNAs which are involved in the processing of pre-mRNA to mRNA, the regulation of gene expression, and the maintenance of telomeres (ends of chromosomes) may show some similarities to the disrupted gene regulation in CHARGE Syndrome (maybe???). And the dysregulation of growth-related pathways in TSC may share some things in common with Lujan-Fryns Syndrome and overactivation of the Shh and Wnt pathways.
As for FXS, well, that one’s a curious cat indeed. But perhaps we’ll find something relevant in next week’s continuation of this several-part series reviewing the basics on syndromic forms of autism.
Ta ta for now!


Pingback: Syndromic Forms of Autism: Part II | Cortical Chauvinism·
Hi, Emily,
Another syndromic form of autism that I would be interested in your take on is the correlation of the oculoauriculovertebral (OAV) spectrum disorders including Goldenhars and hemifacial microsomia. Depending on what study you see, up to 55% of patients on this spectrum also manifest with autism or autistic-like traits. The genetic etiology is somewhat unclear but 14q32 is said to be involved in some cases. I personally have left-sided hemifacial microsomia (involving partial paralysis on the left side of the face, underdeveloped left mandible with skewing of bite and chin alignment, refractive amblyopia of the left eye, and undersized and rounded left ear with chronic tinnitus) plus mild scoliosis but no known involvement of the vital or renal organs. I was recently diagnosed with Asperger’s.
Hi, Pamela. Sorry for the delayed reply. I can’t profess to be an expert in this area of OAV; however, in doing a little lit research I found it highly interesting that there’s a relationship of increased risk for developing OAV and maternal diabetes (Wang et al., 2002). Autism also shares this link with maternal diabetes, so it is tempting to wonder whether, not that OAV causes autism, but that OAV and some forms of autism may instead share a common cause. One of those could be maternal diabetes or there could be other similar factors. Just a thought on my part.