This week is the third and final entry in the series, Syndromic Forms of Autism. It covers an additional five syndromes which exhibit high association with autism. As with the previous blog post, Syndromes II, this includes both single-gene disorders and syndromes derived from duplications and deletions of larger segments of genome spanning multiple genes. In addition, at the end of this entry there will be a listing of additional syndromic forms of autism which have at least a 15% comorbidity rate with autism. In total, including the syndromes covered in previous blogs, there are 45 known high-association autistic syndromes. If you’re interested in learning more, I highly recommend looking them up on Online Mendelian Inheritance of Man or other sources like the Genetics Home Reference.
Smith-Magenis Syndrome
Approximately 90% of individuals with Smith-Magenis Syndrome (SMS) likewise have an autistic spectrum condition. While the greatest disturbances tend to be related to brain function (intellectual disability, language delays, behavioral problems, sleep disturbances, self-injury), individuals with SMS also often have distinctive facial features which become more prominent with age, including a squarish face, prominent lower jaw, down-turned mouth, and a flattened mid-face. In addition to facial anomalies, they also often have hoarse-sounding voices, and sometimes problems with hearing loss. Occasionally, defects in the heart and kidneys are reported.

A young child with SMS. Image borrowed from here.
While the genetic cause of SMS is most typically due to various-sized deletions of the 17p11.2 region which span a number of different genes, occasional cases with single nucleotide mutations in the Retinoic Acid Induced 1 (RAI1) gene have indicated that the primary neurologic, craniofacial, and otolaryngeal symptoms are due to this gene’s loss of function. Meanwhile, other more variable symptoms, such as heart or kidney problems, are probably due to loss-of-function of other nearby genes.
At the present time, it’s uncertain precisely what is the function of the Retinoic Acid Induced Protein 1 (RAIP1), although it is most probably a transcriptional regulator that forms complexes with other proteins and RNA in order to suppress and/or activate expression of target genes. So far, it has been shown to directly regulate transcription of the CLOCK gene, itself a vital regulator in the expression of important genes necessary for proper circadian rhythm functions– the likely reason that sleep disturbances are so common in SMS. Researchers have found that when expression of RAI1 is knocked down in cells, expression of CLOCK proteins are likewise reduced [1]. What other specific targets of RAIP1 exist are currently unknown, though probably have far-reaching effects given the severity of SMS.
Williams-Beuren Syndrome
Approximately 50% of individuals with Williams-Beuren Syndrome (WBS) better known as Williams Syndrome also fulfill criteria for autism. This is a number which flies in the face of the WBS stereotype as people who are overly friendly and talkative, the antithesis to the stereotype of autism. However, it has become apparent that not all individuals with WBS conform to the condition’s stereotype and a significant portion have considerable socio-communicative deficits and stereotypies. All effected individuals, whether typical or not, tend to have mild to moderate intellectual disability and learning disabilities. As for learning disabilities, in particular people with WBS often exhibit poor performance in visuo-spatial tasks and tend to perform better with auditory learning, such as through language and music.
People with WBS tend to have distinctive facial characteristics and can include small chin, prominent lower lip, wide mouth, teeth problems including small, widely-spaced teeth, a long philtrum, a sunken nasal bridge with sometimes a cherub-like nose, stellate or “star-like” patterns within the iris, and puffiness around the eyes. Interestingly, over 80% of the WBS population have either blue or green eyes. They also frequently have cardiovascular abnormalities and sometimes connective tissue disorders, vision problems, issues with the digestive tract, and growth retardation. Increased calcium levels (hypercalcemia) in infancy is also not uncommon.
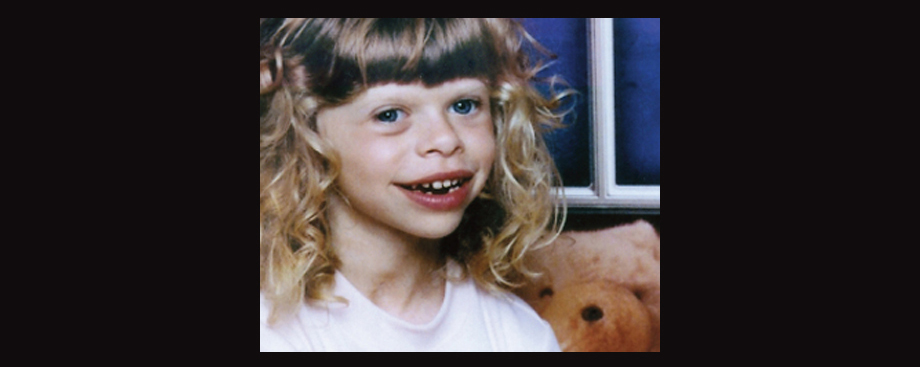
A little girl with WBS. Image borrowed from here.
WBS occurs due to a deletion event in the 7q11.23 region, which spans approximately 28 different genes. While there has been a considerable amount of genetic research performed on WBS, unfortunately this multi-symptom condition has proven complex. The vast majority of individuals with WBS exhibit deletions that include the Elastin (ELN) gene, a major component of elastic fibers in the body whose deletion is especially related to the cardiovascular and connective disorders in the syndrome. However, loss-of-function mutations which target this gene alone do not appear to cause the typical cognitive syndrome of WBS. This may mean one of a few things: either ELN is located nearby the causal WBS gene and is almost always deleted along with it, or the personality profile of WBS is the result of multiple loss-of-function effects spanning two or more genes. Other notable candidates which share this vital WBS region with ELN are LIMK1, RFC2, and EIF4H [2]. LIMK1 likely helps to mediate protein-protein interactions within the cytoskeleton and plays a role in stimulating axonal growth; RCF2 is an accessory protein which plays a role in DNA replication; and EIF4H helps to stimulate protein translation.
Joubert Syndrome
About 40% of people with Joubert Syndrome (JBTS) have autism. Most also have intellectual disability and experience developmental delays. The hallmark feature of JBTS can be seen on structural MRIs, in which the area around the midbrain and cerebellum gives the appearance of a molar tooth on x-ray. This is due to structural abnormalities within the midbrain, the white matter tracts leading to and from the cerebellum, and within the vermis of the cerebellum itself.
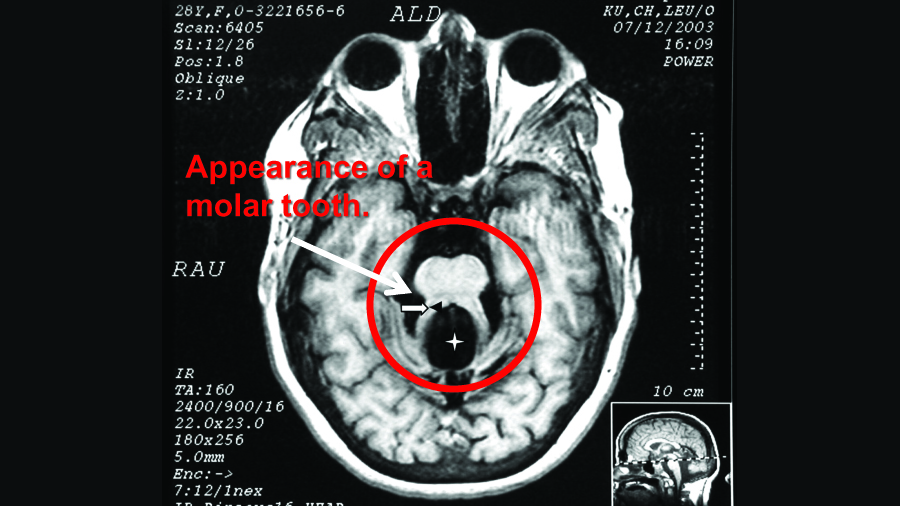
Above is the hallmark “molar tooth” seen in the MRI of someone with JBTS. Adapted and borrowed from here.
Additional features of the syndrome include dysregulated breathing, hypotonia (weak muscle tone) and ataxia, vision disturbances, liver or kidney disease, polydactyly (extra fingers or toes), and endocrine abnormalities.
JBTS is a complex condition, not just because it appears to involve multiple genes but because mutations in a variety of genetic loci can give rise to its core symptoms. Two genes in particular seem to have strong association with the condition, TMEM216 and AHI1, both of which are integrally involved in the development or general function of cilia (hair-like projections) on cells that, for instance, line the ventricles of the brain and serve vital functions.
Velocardiofacial Syndrome
About 28% of those with Velocardiofacial Syndrome (VCFS) fulfill criteria for an autism spectrum condition. Though the functional loss of the TBX1 gene appears to be responsible for many of the core features of the condition, there is considerable symptomologic variability even within families. Common features include heart abnormalities, cleft palate, developmental delays, intellectual disability, and particular facial features. Other features can include microcephaly, hearing problems, short stature, slender hands and feet, and hernias.
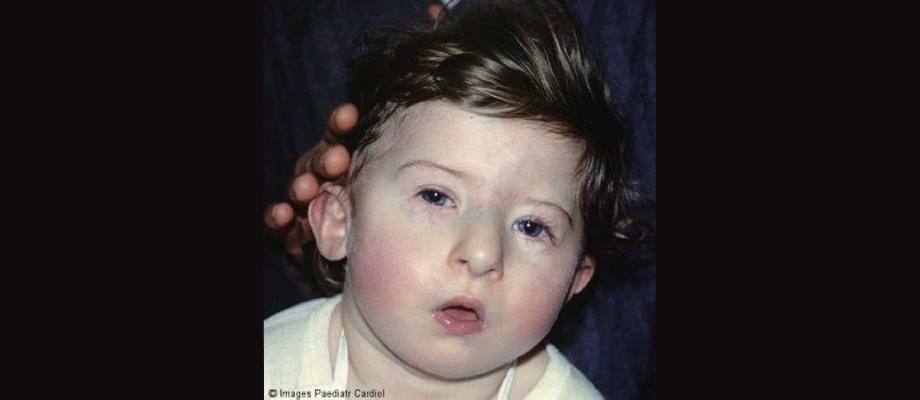
A child with VCFS. Borrowed from here.
As mentioned, the primary gene involved in the etiology of VCFS is T-box 1 (TBX1), which, along with other T-box genes, encodes for a transcriptional regulator necessary for proper development. In addition, separate from its role as a transcriptional regulator, T-box transcription factor 1 also binds and suppresses Smad1, an important player in the BMP pathway. In this role, T-box 1 effectively acts as a BMP pathway suppressor or deactivator [3]. Given BMP’s vital roles in prompting cell differentiation (i.e., maturation from a stem cell-like state), the loss-of-function of TBX1 may suggest that the BMP pathway in effected cells could be over-activated, leading to premature differentiation of cells and an early depletion of the progenitor pool. In mature cells that have already differentiated, BMP overactivation generally leads to poor cell maintenance and disturbed function. (I’ve seen this up close and personal in some of my own BMP-suppressed mouse models I’m working with currently: Well controlled BMP activity is not only necessary for proper tissue development but for continued health and maintenance of adult cells. Some of my mice that overexpress the BMP antagonist, Noggin, in hair follicles rapidly go bald and tend not to regrow much hair.)
Rett’s Syndrome
Rett’s Syndrome (RTT) was once considered an autistic spectrum condition but has now been removed from the DSM5. Instead, it has been recognized that not all individuals with RTT fulfill the criteria for autism and many ultimately outgrow the diagnosis. Nevertheless, approximately 42-58% of those with Rett-typical mutations also fulfill criteria for autism [4]. Almost all cases of RTT occur in females. At one time it was believed this was due to prenatal male lethality but research has instead shown that almost all X-linked RTT mutations occur on the paternal X. Since only females receive the father’s X chromosome, it is almost always females who develop RTT, with a few notable exceptions. Symptoms of RTT are primarily neurological and can include intellectual disability, learning disabilities, communication deficits, microcephaly, loss of motor skills, and repetitive hand-wringing. Most cases of RTT appear to have normal development until around age 6-18 months and then experience a regression, developing signs of the condition. Congenital RTT, however, shows signs at birth and exhibits no regression.

A lovely young lady, named Ariana, who has RTT. Image borrowed from here.
The majority of cases of RTT are due to mutations in the Methyl CpG Binding Protein 2 (MeCP2) gene, which produces a protein product which can bind both DNA and methyl groups, aiding in the expressional “lockdown” of DNA. In order to do so, it forms a complex with a histone deacetylase (HDAC) and SIN3A. Together, these partners suppress gene transcription. MeCP2 appears to be dispensable during the embryonic period but becomes more and more necessary with age, which is a likely cause for the regression which typifies the condition.
What Do These Conditions Have in Common?
Let’s review, shall we?
- Smith-Magenis Syndrome is due to loss-of-function mutations or deletions of the RAI1 gene, whose protein product plays an as-yet-defined role in regulation of gene expression, and in particular appears to regulate production of CLOCK, a protein necessary for proper maintenance of circadian rhythms.
- Williams-Beuren Syndrome is due to a deletion event in the 7q11.23 region which likely includes candidate genes such as ELN, necessary for development and maintenance of elastic fibers in the body; LIMK1, which plays a role within the cellular cytoskeleton and driving axon development; RFC2, an accessory protein in DNA replication; and EIF4H, which stimulates protein translation.
- Joubert Syndrome is related to a variety of mutation events and a number of different genes. Prime gene candidates include TMEM216 and AHI1, both of which share involvement with development or functioning of cilia.
- Velocardiofacial Syndrome is due to a deletion event which includes loss-of-function of the TBX1 gene, whose gene product is both a transcriptional regulator and a suppressor of BMP pathway activation.
- Rett’s Syndrome is usually due to a loss-of-function mutation in the MECP2 gene, a gene product which is involved in the appropriate methylation/deacetylation of DNA and thus gene suppression.
Boy, I tell ya, after three whole blogs of reviewing the etiology of syndromic forms of autism, I have to admit I have more questions than answers at this point. First off, SMS is a hard one since its gene product’s function is so poorly studied, so we’ll just set that one aside for awhile. WBS is especially interesting and holds much possibility, but it’s still hard to say which mutations are indeed the cause of the symptoms (autistic) we’re most interested in. LIMK1 with its effects on axon development? Who knows.
JBTS is tempting, because stem cells such as those which line the ventricles of the brain have cilia which are vital to their function as well proper movement of cerebrospinal fluid (CSF) through the ventricles and the sending of molecular signals through that CSF.
The clearest potential link to other conditions I’ve already covered is with VCFS and overactivation of the BMP pathway, which may be somewhat similar to Lujan-Fryns Syndrome covered in the first blog, which is due to overactivation of the Sonic Hedgehog (Shh) and Wnt pathways following mutations in the MED12 gene. Granted, at face value Shh/Wnt and BMP tend to antagonize one another so on some level they can be considered molecular “opposites”. However, if we’re talking about disruption of cell development, a similar though not identical end can be achieved through overactivating either of these polar opposites. As per example from one of my own studies with hair follicle development, hair loss will ultimately occur regardless of whether BMPs are overactive or Shh/Wnt is overactive. They’re doing slightly different things within the cell, but their cyclical antagonism towards one another is vital for development of the cell. Hence, two opposite paths can lead to the same destination. And while a keratin cell is no neuron, BMPs, Shh, and Wnt have very similar functions throughout all cell types.
And finally, RTT doesn’t seem to have a clear link to the other syndromic conditions I’ve covered. However, disturbed methylation may find links with other forms of autism, specifically in the form induced by prenatal valproic acid exposure (VPA). While VPA doesn’t directly target methylation patterns, it does however directly target histone acetylation through inhibition of histone deacetylases. Studies have found that for the proper suppression of target genes, a combined effort between methylation and histone deacetylation is vital. Without both agencies together, gene suppression is incomplete. Therefore, RTT and prenatal VPA exposure may share some things in common.
And there you have it. Fifteen different conditions with high rates of associated autism, and a HUGE variety of potential causes. Maybe there is a way to whittle these all down to some common root. I know I’ll keep looking and trying– Occam’s Razor after all, right? But maybe… maybe the only commonalities between most of these conditions lie at the level of behavior because we have no definitive biomarkers for autism and our behavioral criteria are so vague that they can potentially encompass an enormous range of syndromes. Is the repetitive handwringing of the little Rett’s girl the same kind of repetitive symptom as the mute little autistic boy who lines up toy cars over and over or the teenage Aspergian girl who memorizes every fact about her favorite teen idol and expounds to any stranger who’ll listen?
Good question. I can’t profess to have an answer. Yet.
Additional Syndromic Forms of Autism
2p15-p16.1 Microdeletion Syndrome
2q37 Monosomy
8p23.1 Deletion Syndrome
WAGR Syndrome
Adenylosuccinase deficiency
Cohen syndrome
Myotonic dystrophy 1
15q13.3 Microdeletion Syndrome
17q12 Deletion Syndrome
Phelan-McDermid Syndrome
7q11.23 Duplication Syndrome
Cri du Chat Syndrome
Epileptic encephalopathy, early infantile, 9
Jacobsen Syndrome
Lissencephaly 1
1q21.1 Duplication Syndrome
XXYY
Klinefelter Syndrome (XXY)
3q29 Microdeletion Syndrome
Cardiofaciocutaneous syndrome
Kleefstra syndrome
Bosley-Salih-Alorainy syndrome
15q24 Microdeletion Syndrome
Duchenne Muscular Dystrophy
XYY Syndrome
PTEN hamartoma-tumor syndrome
Creatine deficiency syndrome, X-linked
5q14.3 Microdeletion Syndrome
Down Syndrome

I’m pretty sure the similarities are mostly at the behavioural level – and even there, it’s often the case that we’re just using similar words to describe quite different behaviours. I’m curious where this 50% figure for Williams syndrome comes from. In my experience (I did a PhD on WS) most people with WS are quite unlike most people with autism. They are, as the stereotype suggests, extremely sociable, although they do have some pragmatic difficulties and often struggle to maintain long-term friendships, perhaps due to their over-sociability. I guess it’s possible that 50% of people with WS could meet the diagnostic criteria for autism, but I think this says more about the diagnostic criteria than it does about WS.
Hi, Jon. Being a n00b to WS I was also quite surprised by the figures I had found. My initial source was an excellent summary article by Betancur on genetic/genomic conditions with associated autism:
http://www.sciencedirect.com/science/article/pii/S0006899310025916
The other which I used for an additional rough estimate was: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0030778
At this point, I’m inclined to agree with your view on the behavioral ambiguity of our autism criteria. Although I do wonder whether some of the genetic syndromes might not adhere better to our classic conception of autism. For instance, I’m curious about individuals with PTEN mutations and macrocephaly, given the larger head circumferences which have been more frequently reported in the idiopathic cases.
I am hopeful that there may be some well-defined genetic autism syndromes which do in fact show the classic syndrome and may give some pointers as to what is occurring within the idiopathic cases. But how to determine that when our measurements are so ambiguous? It’s going to be a challenge.
Pingback: Syndromic Forms of Autism: Part III | Cortical Chauvinism·
Hi Emily that is excellent work on this topic. The genetically determined syndromes have a lot in common there is a high rate of co-occurring and often severe medical problems and a high rate of intellectual disability. What these genetically determined syndromes have in common is that they are almost never inherited. Of the syndromes listed in part 3 only Joubert Syndrome seems to follow some sort of Mendellian inheritance but is quite rare with a prevalence of 1 in 80,000 to 100,000 newborns.
Joubert Syndrome.
http://www.ncbi.nlm.nih.gov/books/NBK1325/
Smith-Magenis syndrome. Virtually all cases are de novo:
http://www.ncbi.nlm.nih.gov/books/NBK1310/
Williams Syndrome: Virtually all cases are de novo:
http://www.ncbi.nlm.nih.gov/books/NBK1249/
Rett Syndrome: 99% of cases are de novo:
http://www.ncbi.nlm.nih.gov/books/NBK1497/
VCSF (22q11 deletion syndrome) 93% of cases are de novo in origin:
http://www.ncbi.nlm.nih.gov/books/NBK1523/
Confounding the association with autism is the work of Howlin & Moss, who found good evidence that in the genetically determined syndromes; autism symptoms are associated with intellectual disability and cautioned against over interpreting the superficial similarities between autism and the behavioral phenotypes seen in most genetically determined syndromes. The authors also found that many genetically determined syndromes have their own
unique pattern of superficial autism symptoms (Moss & Howlin 2009).
For a review of possible mechanisms involved in the production of de novo gene mutations in the genetically determined syndromes you might want review my paper on that very subject:
Click to access 1370118463.pdf
Hi, Robert. Sorry for the delay in response. I’m currently attending a conference and look forward to reading through your paper this weekend and finally respond to your comments. 🙂