Before I get to the autism stuff, I’m going to give you some background. In last week’s blog, I tried to explain the links in development between my hairless mice and the autistic brain. Given some of the feedback, I gather that, well, I did a pretty crappy job of explaining the more difficult concepts which ultimately left people not understanding what in the hades I was on about. So this is kind of a work in progress. This week though I’d like to continue on that theme, get down to more of the basics, and break down for you exactly what I mean by the terms “hyperplasia,” “dysplasia,” and “neoplasia”. After that, I’m going to bring it back to autism, linking these terms with frequent findings in the condition, and why those findings provide strong evidence that most if not all cases of autism have their roots during embryology, which is the first 8 weeks of prenatal life. As a disclaimer before I start, I should state that this doesn’t mean that the severity of autism can’t be affected by postnatal factors.
Okay, so in order to simplify this as much as possible, below is a prototype I’ve put together to represent “normal tissue”. This can be almost any kind of epithelial-like tissue you like; the way they divide and mature are all very similar. In this case, think of this as a prototypical brain, specifically the cortex, with the yellow ovals representing neural stem cells or progenitors and the red ovals representing differentiated or mature neurons. This diagram is highly simplified but it gets to some basic concepts. In this case, each of the second-layer progenitors have produced the neurons for a given column; therefore, the number of columns are reflected in the number of progenitors. Remember this concept for the rest of the following images.
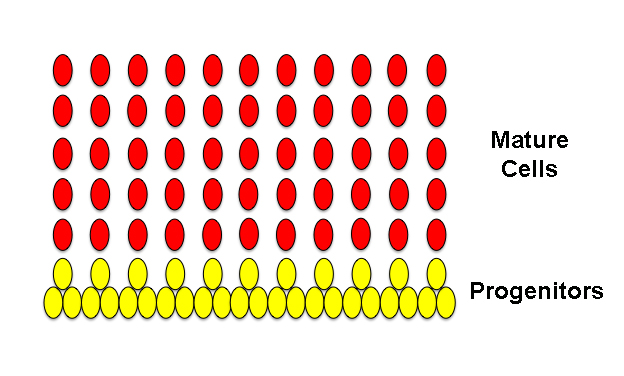
Above, normal tissue.
This next image shows an example of a hyperplasia. At it’s simplest definition, a hyperplasia simply refers to an increase in the number of cells in a given tissue or local area of a tissue. “Hyper” means “over” in Ancient Greek and “-plasia” means “formation“. In the image below, notice that the second-layer progenitors have increased in number, because they have been held in a state of immaturity for a comparatively longer time than in the image above and have therefore continued to produce more progenitors (symmetric division) rather than producing newborn neurons (asymmetric division). Subsequently, since there are more progenitors, we’ve ended up with more columns of neurons.
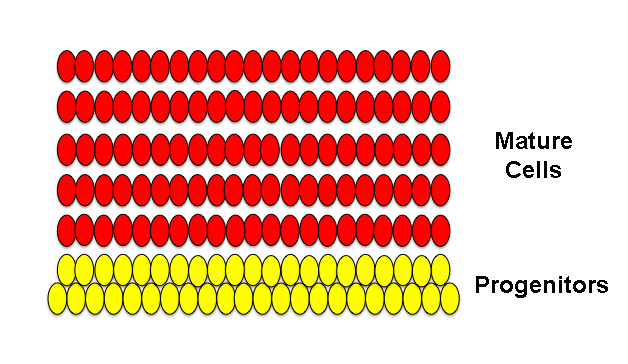
Above, hyperplastic tissue.
Okay, so we have a basic concept: if the progenitors are held in a prolonged state of “symmetric divisions” they continue to produce more progenitor cells, not neurons. In the case of hyperplasia, this effect is temporary and the progenitors go through one or a few extra symmetric divisions but eventually start producing neurons like they’re supposed to. In the case of certain forms of dysplasia, that isn’t the case. In Greek, “dys” means “difficulty” and as we learned above, “-plasia” means “formation,” so that a “dysplasia” is a difficulty in formation. In short, this generally refers to certain types of underdeveloped tissues. In some types of dysplasia, like my hairless mice I was talking about last week, the progenitors continue to be held in a state of symmetric divisions so that the progenitor pool continues to increase in size at the expense of producing neurons. Like the image shows below, you can see that there are more progenitors than in the “normal” tissue but far fewer columns of cells and not as many neurons comprise those columns. (There are different types of dysplasias that can result from different mechanisms of growth retardation, but this is one example and the type which, as you’ll hopefully see later, is the one most relevant to the dysplasias seen in autism.)
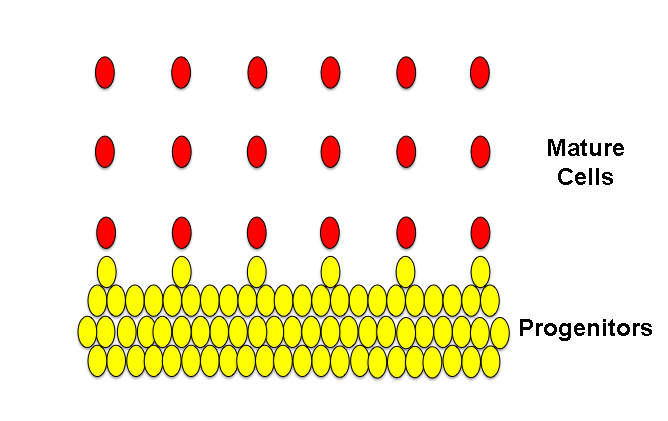
Above, dysplastic tissue.
The last image below is a simple illustration of a neoplasia,, “neo” meaning “new“. In this type of neoplasia, the progenitor cells continue to proliferate, producing even more progenitors and even fewer neurons. As you can see, the cells have grown so much that they’ve formed a bulge. Even though the bulge still shows a certain layered orderliness reminiscent of the normal tissues, some of the flat organization of the progenitor layers is starting to be lost. This is a characteristic of tumors.
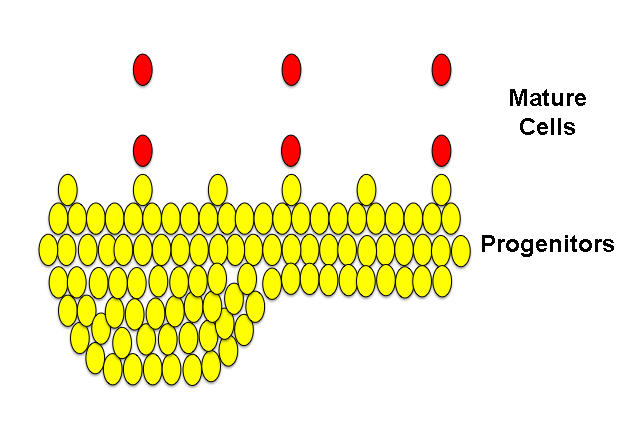
Above, neoplastic tissue.
At this point hopefully you can start to see a common theme: progenitor proliferation. In the case of hyperplasia, progenitors can be held over for a few extra rounds of symmetric mitosis but are eventually able to get back to their normal duties of producing neurons for the developing cortex. This ultimately creates a bigger cortex. In the case of dysplasia, the progenitors are held in symmetric divisions for an even longer period of time and even though they may eventually differentiate and produce neurons, the number and quality of the neurons can be severely affected and the tissue underdeveloped. And finally neoplasia is the most extreme case in which the progenitors don’t switch from symmetric to asymmetric divisions and just continue to produce more progenitor cells. If this continues long enough, the risk of cancer development increases. In these instances, you can look at these three different effects on a continuum of severity, with the length of continued progenitor proliferation underlying the specific phenotype.
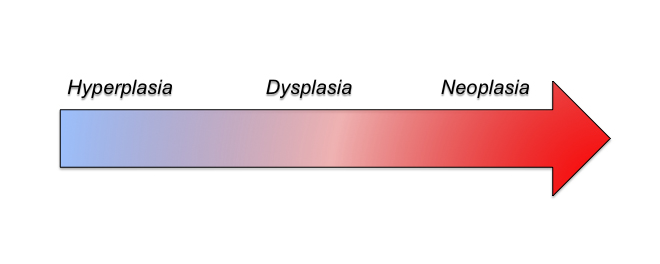
Okay, so now I’m going to switch back to autism. I’ve given you some background on hyperplasias, dysplasias, and neoplasias so that you can understand why the hyperplasias and dysplasias that are extremely common to autism are evidence not only of their inter-relatedness but also that their occurrences support the idea that the roots of autism lie in early cortical development. Around gestational day (GD) 40 is when the developing cortex experiences its first waves of symmetric divisions of progenitor cells. Later, around GD 45 is when these cells begin to asymmetrically divide and produce the first neurons. In the human, neurons continue to be produced for the cortex until around GD 120. In order to develop a hyperplasia, dysplasia, or even a neoplasia within or underlying the cortex, most typically this process must occur during that early period of corticogenesis between GD 40-120. This is also what is seen in the syndromic form of autism associated with Tuberous Sclerosis, in which “tubers” or growths line the ventricles of the brain.
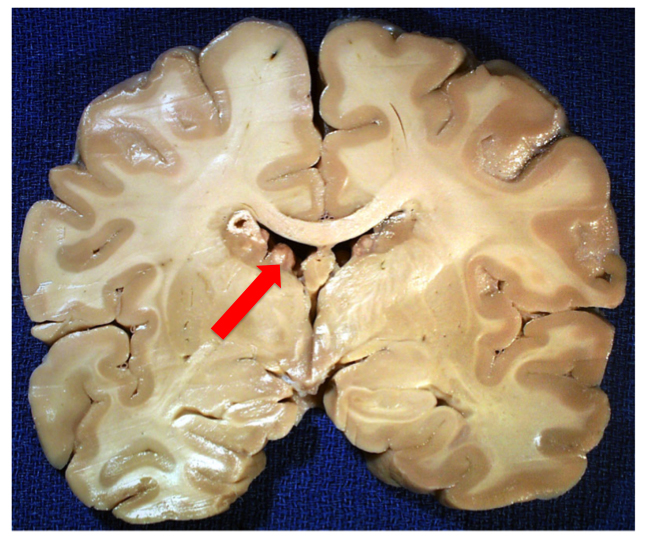
A coronal section of a brain from someone with Tuberous Sclerosis, a syndromic form of autism that is usually a result of a loss-of-function mutation in either the TSC1 or TSC2 genes and which causes upregulation of mTOR pathway activity, leading to increased cell growth and proliferation. In this case, you can easily see the tuberous growths lining the lateral ventricles. Image borrowed from here.
Except in the case of an ongoing neoplasia, the tubers in Tuberous Sclerosis only arise during that GD 40-120 day period. And the same can be said of nonsyndromic autism. And so what do we find in the majority of cases of autistics? We find evidence of hyperplasias in the form of increased minicolumnar numbers, focal dysplasias, and heterotopias, the latter which are neurons which have failed to migrate properly and are located inappropriately in either the gray or white matter [1, 2, 3]. (Because precise timing of proliferation is vital for newborn neurons to receive their proper cues for differentiation and migration, being born at the “wrong time” can lead to miscommunication and the stranding of cells along their developmental and migratory paths.)
In almost 100% of cases of autism, we see secondary features related to affected cell proliferation, notably in the patterns which the gyri (folds) of the surface of the brain take, the size of the gyral window (see image below), and in the size and number of fibers called the corpus callosum that the two hemispheres in the brain send to one another to communicate [4, 5].
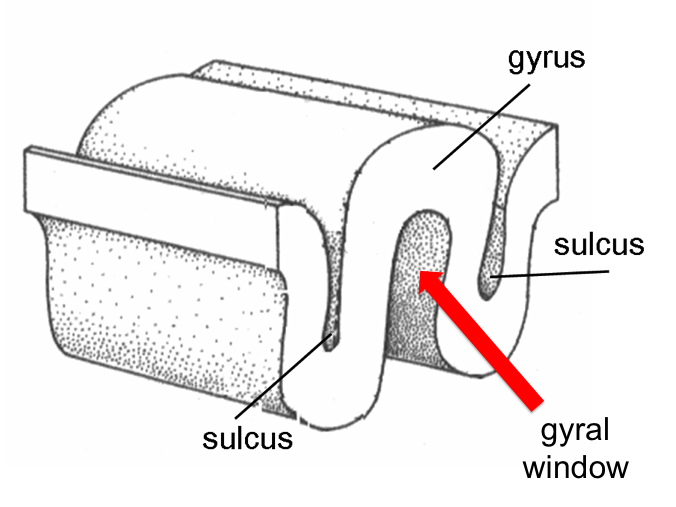
Above is an illustration of the gyral window, which is the volume of space that lies contained within a gyrus.
Specifically, autism exhibits greater gyral complexity, smaller gyral windows, and smaller corpus callosa than their control counterparts. Alongside the evidence of minicolumnar hyperplasias, focal cortical dysplasias, and heterotopias, these all support the theory that the etiology of autism is founded in those very early days of brain development. Irrespective of all the hypotheses and theories on autism out there, the brain doesn’t lie. Pathology doesn’t lie if you know how to read it. And what all of this evidence tells us is that autism begins before the child ever takes a breath.

Hi Emily, do we know if these pathological findings are the same across all ‘types’ of autism – if they are found in children who at first develop normally, then suddenly regress? Even more interested, probably crucial, to know if the same things are found in brains of those who reach ‘optimal outcomes’, i.e. lose their symptoms of autism and regain normal, or nearly normal, neurological functioning? If so it would indicate the ability of the brain to compensate for these structural abnormalities and that they are in themselves not directly linked/causative of what we see as ‘autism’ on the surface…
Btw could the findings of abnormal lung branching in autism be somehow related to what you are seeing in the brain? http://www.ncbi.nlm.nih.gov/pubmed/22926922
Some background here http://thorax.bmj.com/content/55/7/587.full
Hi Emily –
Super great post!
1) I love the nuance that is so dreadfully missing from so many discussions on causation; Very nice.
2) Your diagrams are very helpful for a simple drone like myself, and I imagine, for others as well. I have previously tried getting through some of the denser papers on this subject, including a few by Dr. Cassanova, and found that my spatial modeling skills just weren’t there to visualize the perturbations of neuronal migration patterns in three dimensions [with folding!]. This is definitely a good start.
Regarding hyperplasia, which seems to have findings in the autism arena, I am curious on your thoughts on possible (?) participation of a *failure* to ‘prune’ the system later in life as a factor. [and by later, that can still mean in utero.]. For example, the Courchesne paper on increased neuron numbers in autism indicated that *usually* many of the ‘excess’ neurons are culled in the last trimester of pregnancy and postnatally, and that it was possible that a failure of this process could be part of the reason of increased neuron numbers. This would seem to be plausible with the recent findings of altered microglial phenotype in the autism population and observations that those same cells participate in pruning and synaptic maintenance.
Unfortunately, I’m far too behind in neurobiology to understand how much (if any) ‘pruning’ happens in the areas of observed hyperplasia in the autism population. Can you help?
Thanks!
– pD
Hey, PD. Thanks for the compliments and glad the diagrams were helpful!
In regards to pruning, I could definitely see a lack-of playing a role and perhaps in a minority of cases this could be the case. But as far as explaining the bulk of the neuropath data, problem-pruning wouldn’t explain the dysplasias or the heterotopias. So I think it’s a feasible hypothesis that may well turn out to be accurate for a subset of autistic people, but the majority probably have issues with prolonged proliferation of the progenitors, which would be the primary means for creating dysplasias and ill-migrated cells. Does that make sense?
Hi, Natasa. I had the exact same questions as you but unfortunately when it comes to various neuropathological postmortem studies, regression invariably seems to be an exclusion factor. Disappointing I know, so that unfortunately leaves that question unanswered. But it is something I hope that we can get to answering. With regards to your other question, the studies I mentioned did include higher-functioning individuals, although I suspect that they still needed to currently fulfill diagnostic criteria in order to be included in the MRI studies. The postmortems did include some adults but was heavily represented by children, so a bit too young to tell there probably. But I agree, that’s likewise an important study to perform to see whether these disturbances are present in all range of autistic– or perhaps even Broader Phenotype– individuals. I would hesitate to say however that if it’s present in every case that it has little to do with the outcome of phenotype. Remember that these effects can be seen on a continuum. Whereas the hyperplastic effects may provide for higher intelligence and focal talents, the dysplasias are unlikely to prove quite as potentially beneficial. One thing which I would like to be addressed in future is whether there are links to the severity of the autism and the number of occurrences of dysplasias and heterotopias. I hypothesize that there may be more of these occurring in those much more severely affected and also the nature of the underdeveloped tissue may be substantially more disorganized. There have been numerous studies linking the occurrences of cortical dysplasias and heterotopias to the seat of seizure development. So the frequency of seizure disorders in autism may not be especially surprising with this knowledge.
As far as the findings for bronchial branching in autism, that sounds highly relevant and may provide further evidence of a multi-tissue effector that targets a given time period of development (embryo/early fetal) and affects not only brain growth but other systems which are developing at that time. Frankly, if we’re talking about an exogenous agent, a body-wide endogenous molecule, or a gene that’s actively expressed/repressed during this time period in multiple systems, you will expect to see many tissues affected, even if only subtly. Autism has the potential, case by case, to be syndromic and not just a condition of the central nervous system. Thus far, the primary thing that the brain and the lungs have in common is that they’re epithelial tissues and therefore may be vulnerable to similar effectors in similar ways. The gut can also be included in this and I’m still curious whether on closer histological inspection there may not be signs of GI developmental disturbances.
Thanks Emily! Apart from the gut, how about abnormal folding/inclusions in the placenta found in autism pregnancies? http://www.ncbi.nlm.nih.gov/pubmed/23623455
That should predate the time period we’re talking about, but that doesn’t preclude it from happening. If there’s an endogenous/exogenous stressor, it may be quite capable of leaving its telltale mark on multiple stages of development. For instance, my interest in de novo mutations would arise much earlier than neocortical development, although the two may still be related by a common stressor.
In addition, I suspect some cases of abnormal placental development could be related to endocrine issues which have found links with autism (e.g., diabetes during pregnancy). Iirc, fertility issues and eclampsia have links with troublesome maternal-placental formations and interactions.
On this note http://sfari.org/news-and-opinion/conference-news/2013/society-for-neuroscience-2013/maternal-infection-may-alter-neuronal-signals-connections/
Thanks, Natasa, very interesting. Although, once again, ironic that all the focus is placed upon the synapse and neuronal proliferation is not even considered. But I suppose that’s the big thing in autism right now, the synapse. But cytokines/chemokines have certainly been shown to alter proliferation.
@Emily –
Well, this guy from SFARI indicates problems with unchecked proliferation as a component, so someone out there is paying attention!
http://sfari.org/news-and-opinion/conference-news/2013/society-for-neuroscience-2013/neurons-made-from-people-with-autism-show-distinct-markers
Take care.
Hi, PLD. Thanks for the link. I wasn’t aware of that particular in vitro study, but am definitely familiar with more of Flora’s work on FGF in autism. She’s definitely a proponent of the work I’ve cited above and she is a friend of Manny’s. The results regarding GABAergic neurons are unclear; however, in vitro studies are always rather suspect in regards to aspects of cell development– especially that of cell identity, since so much regarding identity is determined by the local tissue environment and can rarely be reproduced appropriately in a petri dish. (I can’t tell you how many different studies I’ve read that’ve reported in vitro production of a certain cell type under an exogenous agent, and then when a mouse model is made to reproduce it, the exact opposite occurs in life!) Also, they remarked on the age of the cells and there may be characteristics of differentiation that remain from the skin cells which drive these “neurons” towards an inhibitory profile. But highly interesting, thanks!