“How a genotype and its environment interact to yield a phenotype poses a vast epistemological gap. Proteins that exhibit conformational diversity and contingent functional multiplicity increase the dimensions of phenotypic space encoded by any given genome. Protein folding can therefore radically alter the trajectories that connect genotype and phenotype, modify adaptive landscapes and influence evolution” (p. 435, Shorter & Lindquist, 2005).
Perhaps you’ve heard the adage, “Shape determines function”. If not, think about how the shape of a cup enables you to drink water more easily. Or how the shape of a chair assists you to sit. These are simple analogies for comparison to the molecules of a cell, but they are nevertheless instructive ones. A given molecule, be it a string of nucleotides, carbon atoms, or amino acids, is not just a sequence of blocks or letters (A, T, C, G). They are 3-dimensional objects whose shapes change dynamically dependent upon their chemical partners. And those shapes determine their functions.
I’m sure most of us remember Mad Cow Disease. Otherwise known as a variant of the Creuzfeldt-Jakob Disease, many cows across the world had been infected with this agent, which was later determined to be a misfolded protein called a prion. The United Kingdom was hardest hit by the infection, necessitating the euthanization of approximately 4.4 million infected cattle. It is believed that the infection may have originally come from a similar disease in sheep known as Scrapie. Cows were fed a meat and bone meal comprised of previous cows’ remains but which had also been processed on the same machinery as infected sheep. The misfolded proteins were ingested, leading to infection and subsequently to the spongiform encephalopathy that produced fatal neurodegeneration in the affected animals.
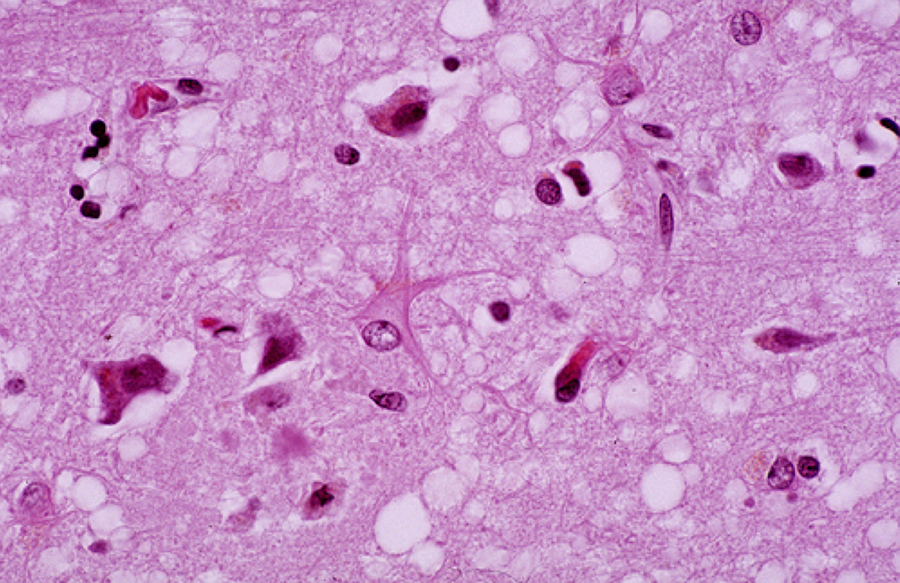
An example of Spongiform Encephalopathy as would be seen in Mad Cow Disease. Image borrowed from here.
What is a prion and how is it potentially infectious? A prion is simply a native protein which is misfolded. Beyond this, it has the capacity to bind to other proteins which usually share a similar amino acid sequence to the prion and induce their misfolding. In short, it acts like a dangerous domino effect, promoting the malformation of target proteins which can then cause problems for the cell, not only depleting the cell of that protein’s function but also causing a build-up of protein waste in the form of inclusions. (You may remember the FMRP inclusions which were mentioned in relation to Fragile X Tremor/Ataxia Syndrome (FXTAS) a couple weeks ago.) Eventually, such inclusions have the potential to destroy the cell in which they reside.
At one time it was believed that the aggregation of inclusions in various conditions such as the tau inclusions of Alzheimer’s were cell autonomous (i.e., protein misfolding occurred independently in each individual cell). However, it’s more recently been shown that the passage of prion-like proteins from cell to cell also probably plays important roles in the spread of these disease states across a given tissue such as the brain [1]. Therefore, prions, though having made a huge splash in the media with Mad Cow Disease and slowly falling out of the lime light as if an anomaly, may play subtler and more sinister roles in diseases very well known to us.
But are prions always pathological? Could these prion-induced pathologies simply be the unfortunate byproduct of a more normal state of complex biological affairs? As Shorter and Lindquist suggested in 2005, perhaps prions share some capacity for non-genetic inheritance with their impressive capacity to alter the shape, and therefore the function, of other proteins. This could be passed down from one cellular generation to the next. After all, even a newly conceived zygote hasn’t been made “from scratch”. True, some patterns are wiped clean and started afresh, but others still remain. The proteins present within the egg itself are all inherited, as are the fats, the carbohydrates, and other inorganic substances. But even if we’re only talking about lateral inheritance, or the passage of a replicative element from one neighboring cell to the next, prions could still play huge roles in general development. As Shorter and Lindquist write:
“Prion conformers operate as a template for other conformers, usually of the same amino-acid sequence, to acquire the prion conformation, and these, in turn, are templates for others, creating a protein-folding chain reaction. This self-replication of conformational information enables prions to act as genetic elements with the ability to transmit disease, encode heritable phenotypic traits or encrypt molecular memories. That prions are a conduit for the replication of heritable information places them in the codical domain, more commonly regarded as the territory of DNA or RNA, and potentially empowers prions to operate as units of selection.”
Inclusions which result in diseases like Alzheimer’s and FXTAS may be indicators of when the normal prion-like function of certain proteins has gone awry. Interestingly, many prion-prone proteins are rich in the amino acids, glutamine and asparagine. Perhaps then it is unsurprising that a number of trinucleotide repeat disorders exhibit amplified numbers of glutamine repeats within their coding sequences. These conditions include Huntington’s Disease, spinobulbar muscular atrophy, and many of the spinocerebellar ataxias. Each of these poly-glutamine disorders exhibit neuronal inclusions akin to those seen in prion-induced conditions like mad cow disease [2]. Although not all prions are rich in these amino acids, it is however a considerable trend.
Ultimately, studying the behaviors of target amino acids like glutamine may help us understand not only the pathologies that may arise from them, but how they have evolved to behave in the first place. Perhaps amino acids such as glutamines and asparagines have been selected for for these particular qualities, and that, amongst other functions, they are purposeful inducers of conformational– and perhaps even inherited– change.


{kind=link}