For most people, if they’re at all familiar with Ehlers-Danlos Syndrome (EDS) or the closely related Hypermobility Spectrum Disorders (HSD), when they think of these conditions the first thing that comes to mind tends to be something along the lines of “connective tissue.” Perhaps they may envision someone who can bend his/her body into impossible positions or who has stretchy skin. But not as many people realize that EDS/HSD usually has a whole host of comorbid issues that, at first glance, seem to have little to do with connective tissue and yet for some people can be even more impairing than the joint instability and musculoskeletal pain itself.
Every person with EDS/HSD faces overlapping but unique daily challenges. For some people, gastrointestinal disorders can be especially impairing– even debilitating. Some EDSers may barely remember what it’s like to go with family and friends to restaurants because they’re so sensitive to most foods that they’ll inevitably end up horribly sick. Some may face the torture of perpetual gastroparesis in which their “normal” is throwing up several times a day after meals. And others still may be so constipated that they only go to the bathroom every other week and live off laxatives or even enemas.
Although not everyone with EDS/HSD has gastrointestinal (GI) problems this extreme, these issues are nevertheless very common in this population. For some, GI disorders like ulcers may be an inevitable risk with the chronic use of NSAIDs to treat the extreme musculoskeletal pain that often goes along with the condition. For others, the connective tissue laxity, itself, may predispose towards various prolapses and hernias that can cause a physical obstruction of the GI tract. But most people on the EDS/HSD spectrum have functional GI disorders (FGD), which are rooted in dysregulation of the autonomic nervous system (ANS) that coordinates the activity of the gastrointestinal tract. Sometimes it may be overactive as in the case of “dumping syndrome” and diarrhea, and other times it may be sluggish, as in gastroparesis and constipation. And many times it fluctuates over time for an individual– even throughout the same day!
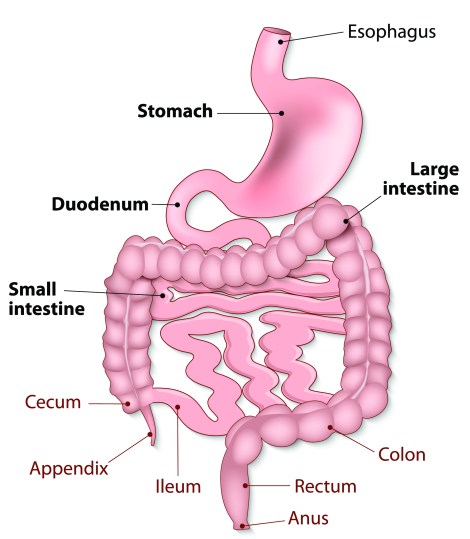
While it’s not certain what leads to functional changes in the ANS, given the chronic inflammation and tissue damage we experience, progressive neuropathy (nerve damage) is one possibility in more severe cases of FGD, especially as the extracellular matrix (e.g., collagen) is vital for repair of the ANS. When a nerve cell in the ANS is damaged, connective tissue forms a structural and communicative platform along which that nerve can regrow. And so for someone with weak collagen like us zebras, it’s feasible that that repair process may not work well. Therefore, when inflammation leads to damage of the nerve fibers surrounding the GI tract, our bodies may not be great at repairing them, ultimately leading to progressive changes in the way the ANS functions and regulates the GI.
On the other hand, many people on the EDS/HSD spectrum also seem to have lifelong problems with dysautonomia, which refers to a general dysregulation of the ANS. This can lead to GI issues, as well as other problems related to orthostatic tolerance, fatigue, etc. So it’s also possible that although some people with extreme FGD may have some neuropathy, most zebras may simply have a hereditary propensity for our ANS to be a little wonky. That may be due to actual dysfunction of the nervous system itself or the ANS may be responding to dysfunction within another system of the body with which it interacts, such as the immune system. Links between conditions like postural orthostatic tachycardia syndrome (POTS) and mast cell activation syndrome (MCAS) suggest the latter may sometimes be the case. In addition, my group’s ongoing research into the links between autism and EDS/HSD also suggest there’s strong ties between the immune and autonomic nervous systems. GI disorders are also very common in the autism spectrum. (More to come on those topics soon!)
So anyways: in fine zebra fashion, we’re complicated. Zebras of many colors. Some of us have minimal GI distress, others may be mildly or moderately impaired, and still others of us may be completely debilitated by GI disorders. Like all of our other issues, it varies person-by-person, day-by-day, and sometimes (*sigh*) even hour-by-hour.

A special thanks to all the zebras on the Ehlers-Danlos Support Group on Facebook who took the time to offer their experiences to me concerning their gastrointestinal disorders. This post is for you. ❤

I forgot to share this with you…
This is from the article Neuromuscular and Systemic Presentations in Adults: diagnoses beyond MERRF and MELAS..”Mitochondrial dysfunction plays a central role in both aging and muscle deconditioning from disuse, but this misses the point regarding pathogenicity. The second point is that myopathy may occur in persons with Ehlers–Danlos syndromes (EDS), and the overlap in symptoms in EDS and mitochondrial disorders may make it difficult for the clinician to distinguish these disorders. Infants and young children with EDS may be labeled as hypotonic, whereas older children and adults will report weakness and easy fatigability. In EDS and other hypermobility disorders, the patient must first exert force to stretch the tendon and keep it taught before the belly of the muscle can shorten to move the joint against the resistance. Furthermore, muscles surrounding the joint that are not even involved in the primary intended movement must contract across the joint in all planes of freedom in order to stabilize the joint. This extra work is required with every movement and creates perceived loss of muscle strength, early fatigue, and pain. There is also evidence that the maximal extensional weakness is caused by excessive compliance of the series-elastic component of muscle tissue [24]. There is also evidence that there is loss of proprioception, at least in the ankles, which leads to gait instability [25]. In one series 85 % of patients had mild or moderate muscle weakness, 60 % had reduction of vibration sensation, 13 % had an axonal polyneuropathy, and 26 % had myopathic electromyography findings. Muscle atrophy occurred in half of patients with myopathic findings on muscle biopsy of 28 % [26]. Although the degree of muscle symptoms vary for patient to patient, true muscle weakness is a feature seen even in patients with the least pathogenic forms of EDS. What cannot be explained is that some people with hyperextensible joints have no perceived weakness and are quite athletic. Another common feature in many EDS spectrum disorders is bowel dysmotility and dysfunction, a feature also found in those with mitochondrial disorders. The dual features in EDS of weakness and bowel dysmotility create an symptom set common in mitochondrial patients. Creatinine kinase (CK) is generally not elevated in EDS, but this is of limited usefulness because CK is not elevated in many cases of mitochondrial myopathy. The protean nature of EDS and it’s neuromuscular similarity to mitochondrial disorders suggests that this disorder must be considered in persons undergoing any neuromuscular evaluation, including a mitochondrial evaluation”.🦓
YA RIGHT..NEURO DID NOTHING AND DISMISSED WHEN I SUGGESTED SOME MITO INVOLVMENT.